



Okay, here we go. If I may, letters (and anyone else who can chime in) I'd like to ask some questions and clarify some things, line-by-line. That could be the easiest way here.
take 74.27g of methylamine.hcl (1.1mol), dissolve in the minimum amount of water and write it down.
add toluene (5-10 times the amount of water) then drop in 1mol of NaOH flakes (40g), slowly while in an ice bath.
I'm going to eliminate the ambiguous spread here, and just say 10x the amount of water, is that okay? And this could use stirring, right? You mention shaking, but not stirring (shades of James Bond). I'd like to do this all with clamped, set-up glass if possible.
And the ice bath mentioned, that can be set up in advance, correct? In other words, that first step of dissolving the amine in water can be done in glass that's already in said ice bath, right?
then add more NaOH, 6-8g per every ml of water present. So if you had to dissolve your amine in 50ml water, you must add 300-400g dry NaOH.
Again, the "6-8" is ambiguous to me as a layman; I have no idea why to choose one end of that spectrum over the other. Can we just go w/the maximum, and say 8g per mL?
Then add 150g more NaOH to account for the water formed by the reaction between OH(-) and MeNH3(+). Shake well and let sit in the ice bath till everything settles down.
Please describe what it was doing that it now has to "settle down." Exothermic action? Mild bubbling? Heavy frothing? I will need to know in advance. I'm also wondering again about stirring vs shaking. If this is a fixed glass setup, I'd rather be stirring, but maybe this is too much solids to be magnetically stirred...? Maybe it *has* to be swirled by hand? Those are the kinds of physical details I need to get a grip on. (Those ratios look to me like there's enough liquids that it could all be magnetically stirred, but I'm not sure)
If there isn't even any need for a condenser or addition funnel, I guess we aren't talking about any kind of clamped glass setup, just a flask on a stirplate, right? If it's going to be mag-stirred at any point, I can't pick it up and shake/swirl it because there will be a stirbar in there (unless I pull it out, of course). These kinds of aspects are obvious to the seasoned pro, but for someone like me, every little detail of this type has to be known in advance. And are we sure there's no need for a more anhydrous setup, like having all the flask necks stoppered except for one, with a drying agent plug in it?
Decant or filter your toluene, add to it some methanol and 0.9mol of your ketone, stir 30 minutes, then add 10~17g NaBH4 (depending on quality of NaBH4) in small portions in the ice bath, let stir extra 2 hours @ r.t. after addition of NaBH4 is done, and workup as usual. This works and is easy!
It's actually the whole reaction mixture that I'm decanting or filtering at this point, right? You say "decant the toluene" but I assume you really mean the entire mix since it is all intimate at this point. And what am I decanting away from---undissolved NaOH?
"Some" methanol is too vague, I'm afraid. Please give an amount, or an objective I can relate to (i.e. is the point to add enough MeOH to make the mix more easily stirrable?)
It will definitely be the greater amount of NaBH4 in my case because it is old NaBH4. I take it that it's added straight (dry), in small portions due to reactivity? It does visibly react, correct ? Maybe you could suggest something more specific as a way to add it (example "add portions of NaBH4 at a rate of roughly 0.5 g every 15 seconds until 17g has been added" or something like that). In the end though it will be more than 17g because we're going to scale this up to 100g ketone if we can.
If you will be kind enough to answer these questions (and help me adjust scale/amounts/moles to 100g ketone) I will then have more than enough to write up a very nice method!
take 74.27g of methylamine.hcl (1.1mol), dissolve in the minimum amount of water and write it down.
add toluene (5-10 times the amount of water) then drop in 1mol of NaOH flakes (40g), slowly while in an ice bath.
I'm going to eliminate the ambiguous spread here, and just say 10x the amount of water, is that okay? And this could use stirring, right? You mention shaking, but not stirring (shades of James Bond). I'd like to do this all with clamped, set-up glass if possible.
And the ice bath mentioned, that can be set up in advance, correct? In other words, that first step of dissolving the amine in water can be done in glass that's already in said ice bath, right?
then add more NaOH, 6-8g per every ml of water present. So if you had to dissolve your amine in 50ml water, you must add 300-400g dry NaOH.
Again, the "6-8" is ambiguous to me as a layman; I have no idea why to choose one end of that spectrum over the other. Can we just go w/the maximum, and say 8g per mL?
Then add 150g more NaOH to account for the water formed by the reaction between OH(-) and MeNH3(+). Shake well and let sit in the ice bath till everything settles down.
Please describe what it was doing that it now has to "settle down." Exothermic action? Mild bubbling? Heavy frothing? I will need to know in advance. I'm also wondering again about stirring vs shaking. If this is a fixed glass setup, I'd rather be stirring, but maybe this is too much solids to be magnetically stirred...? Maybe it *has* to be swirled by hand? Those are the kinds of physical details I need to get a grip on. (Those ratios look to me like there's enough liquids that it could all be magnetically stirred, but I'm not sure)
If there isn't even any need for a condenser or addition funnel, I guess we aren't talking about any kind of clamped glass setup, just a flask on a stirplate, right? If it's going to be mag-stirred at any point, I can't pick it up and shake/swirl it because there will be a stirbar in there (unless I pull it out, of course). These kinds of aspects are obvious to the seasoned pro, but for someone like me, every little detail of this type has to be known in advance. And are we sure there's no need for a more anhydrous setup, like having all the flask necks stoppered except for one, with a drying agent plug in it?
Decant or filter your toluene, add to it some methanol and 0.9mol of your ketone, stir 30 minutes, then add 10~17g NaBH4 (depending on quality of NaBH4) in small portions in the ice bath, let stir extra 2 hours @ r.t. after addition of NaBH4 is done, and workup as usual. This works and is easy!
It's actually the whole reaction mixture that I'm decanting or filtering at this point, right? You say "decant the toluene" but I assume you really mean the entire mix since it is all intimate at this point. And what am I decanting away from---undissolved NaOH?
"Some" methanol is too vague, I'm afraid. Please give an amount, or an objective I can relate to (i.e. is the point to add enough MeOH to make the mix more easily stirrable?)
It will definitely be the greater amount of NaBH4 in my case because it is old NaBH4. I take it that it's added straight (dry), in small portions due to reactivity? It does visibly react, correct ? Maybe you could suggest something more specific as a way to add it (example "add portions of NaBH4 at a rate of roughly 0.5 g every 15 seconds until 17g has been added" or something like that). In the end though it will be more than 17g because we're going to scale this up to 100g ketone if we can.
If you will be kind enough to answer these questions (and help me adjust scale/amounts/moles to 100g ketone) I will then have more than enough to write up a very nice method!
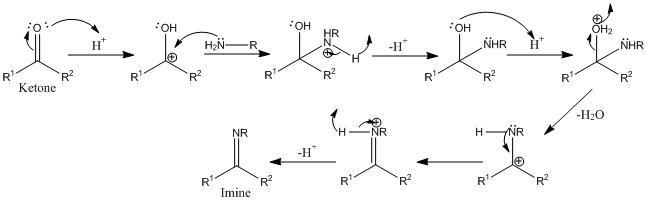



 ). I wrote it at the 100g ketone scale, since that's where I want to run it ultimately, but when I go to do it the first time I will probably scale it down 90% (10g ketone) as per your recommendation.
). I wrote it at the 100g ketone scale, since that's where I want to run it ultimately, but when I go to do it the first time I will probably scale it down 90% (10g ketone) as per your recommendation.

