Abstract
The direct oxidation of codeinone (5) to 14-hydroxycodeinone (6) was investigated with a wide range of oxidizing agents. The majority of reagents gave little or no desired product. Peracids were found to be moderately successful, with dimethyl peracetic acid giving the greatest yield (37%). Metallic oxidants generally gave rise to competing oxidations, however Co(AcO)3 was found to conveniently oxidize 5 to 6 in 51% yield, representing a practical, non-chromatographic procedure for the preparation of 14-hydroxycodeinone.

The 14-hydroxyl substituted opioid antagonists, such as naltrexone (1) and naloxone (2), are valuable medications for the treatment of opiate abuse,1 opiate overdose,2 and alcohol addiction.3 However, the ever increasing demand for these medicinal opioids, coupled with the findings that delta subtype selective derivatives may be useful immunosupressants4 and also prevent the development of tolerance to morphine,5 has placed a premium on thebaine (3), the common starting material6 that has only a low natural abundance in opium.7
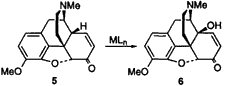
Obviously, the replacement of 3 as the starting material with a more available opiate, such as codeine (4) would alleviate the pressure on thebaine. As part of our program directed toward this goal, we recently reported a novel and practical synthesis of 3 from 4.8 Codeinone (5), which can be prepared through simple Oppenhauer oxidation of 49, was shown to undergo enol ether formation to 3 by treatment with tBuOK, 18-crown-6, and Me2SO4, thus removing the need for the isolation of 3 from opium. Peracid oxidation of 3 gives the key intermediate 14-hydroxycodeinone (6),10,11,12 from which the important antagonists can be prepared.6 We now wish to present our studies into the oxidation of 5 at the allylic position to give 14-hydroxycodeinone (6) directly, and thus remove the requirement for the preparation of thebaine as an intermediate in the syntheses of the antagonists.

The oxidation of 5 to 6 can be accomplished by a variety of 2-step procedures via formation of a diene system followed by oxidation.13,14 Obviously, a direct procedure would offer the advantages of less synthetic steps and remove the necessity for the isolation of a diene intermediate. Previous attempts at such a direct oxidation have met with limited success due to competing oxidation at other positions on the opioid skeleton.14,15 The only direct method resulting in a reasonable yield involves the use of MnO2;16 treatment of codeine with MnO2 leads to codeinone, however prolonged treatment gives 6 in 30% yield. Although this procedure was subsequently improved using a 3-silyl ether derivative (59%)17, the large quantities of MnO2 employed limits its practicality for industrial scales. In an attempt to develop a practical method for this transformation, we investigated the treatment of 5 with a variety of readily available and easily handled oxidizing agents, with the ultimate aim of discovering a procedure that could be conveniently performed on commercial scales.
Peracid Mediated Oxidations
Initial studies concentrated on the fact that in acidic media, codeinone (5) exists in equilibrium with neopinone (7).18 It was envisaged that the different electronic character of the C-ring double bonds in 5 and 7 may allow selective oxidation of 7 with peracids, whereas the electron deficient double bond in 5 would not undergo oxidation. It was found that treatment of an equilibrium mixture of 5 and 7 under the performic acid conditions described by Iijima et al. for thebaine,10 led to low yields of 6 (Table 1, entry 1).
Oxidation of 5 with peracids (Table 1)

Although somewhat encouraging, the fact that the balance of the material could not be recovered suggested competing oxidations to give water soluble products. A range of peracids, with differing steric and electronic properties were therefore investigated in order to determine if the nature of the peracid affected product distribution and the results are summarized in Table 1.
| Reagentsa | Yield of 6 | |
1 | HCO2H/H2O2, 40°C, H2SO4 | 10%b |
2 | HCO2H/H2O2, H2SO4 | 13% |
3 | HCO2H/H2O2, HClO4 | 9% |
4 | F3CO2H/H2O2, H2SO4 | |
5 | H3CCO2H, H3CCO3H, H2SO4 | 5% |
6 | m-CPBA, H2SO4 | 4%d |
7 | m-CPBA, F3CCO2H | |
8 | Cl2HCCO2H/H2O2, H2SO4 | 5% |
9 | Cl3CCO2H/H2O2, H2SO4 | |
10 | (CH3)2HCCO2H/H2O2, H2SO4 | 37% |
11 | (CH3)3CCO3H, H2SO4 |
- All reactions stirred for 24h at rt,
unless otherwise stated; - Conditions as described by
Iijima et al. for thebaine10 - No desired product detected by TLC
- Reaction performed in water as
done by Coop et al. for oripavine12 - As entry 6, but in CF3CO2H solvent.
- Peracid prepared as described.19
- Reaction performed in t-BuOH.
As can be seen from the table, all reactions gave rise to only minor yields of 6, the greatest yield (37%) was obtained by the use of dimethylperacetic acid (entry 10). The low yields make it difficult to draw any definitive conclusions, however the fact that dimethylperacetic acid gave a higher yield than peracetic acid (entry 5) tends to suggest that steric bulk may be one factor in minimizing unwanted side reactions. This however, is not consistent with the poor results obtained with the even more bulky tri-substituted peracetic acids (entries 9 and 11). In all reactions the side products were water soluble and could not be isolated.20 Although strong acid was included in all cases, the possibility of N-oxidation as one side reaction cannot be dismissed,21 and therefore the action of peracids on the corresponding carbamate protected derivative (8)8 was investigated (Table 2).
| Reagentsa | Result | |
| 1 | HCOOH/H2O2, H2SO4 | |
| 2 | F3CCO2H/H2O2, H2SO4 | |
| 3 | H3CCO2H, H3CCO3H, H2SO4 | |
| 4 | (CH3)2HCCO2H/H2O2, H2SO4 |
a. All reactions stirred for 24h
at rt, unless otherwise stated
b. Complex mixture of oxidation
products, no desired
product detected by TLC.
Oxidation of 8 with peracids (Table 2)

Surprisingly, treatment of 8 with peracids led to none of the desired 14-hydroxyl substituted product (10). This is however consistent with previous findings that basic and non-basic opioid derivatives can react very differently when treated under the same conditions.22 Little starting material could be recovered in any of the reactions and it was concluded that alternative oxidations (possibly Baeyer-Villiger20) competed successfully.
Metallic Oxidants
Figure 1
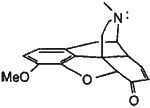
The possibility that Baeyer-Villiger reactions of 7 and 9 were successfully competing with the desired oxidations above, suggested that the optimal reagent for this transformation will be one that is not capable of performing a Baeyer-Villiger reaction. In addition, it was concluded that some method of regiocontrol may be necessary to favor the desired oxidation over benzylic oxidation at C-10.23 As it has been shown that the N-lone pair of opioids occupies a position above the C-ring,24 it was considered that an oxidizing metallic species coordinated to the N-lone pair of 5 may be suitably positioned close to the desired site of oxidation thus favoring oxidation at this position. (Figure 1).
Encouraged by the recent report that a metallic species coordinated to a polyaniline ligand together with a cooxidant gave rise to allylic oxidation,25 we decided to investigate the application of such a procedure to 5, where the opioid tertiary amine could act as the amino ligand. Thus 5 was treated with a range of readily available metallic oxidants, and the results are summarized in Table 3.
| Reagentsa | Yield of 6 |
|
| 1 | FeCl3 | |
| 2 | Co(OAc)2 cat., NaIO4 | 12%c |
| 3 | Co(OAc)2 cat., Oxone | |
| 4 | Co(OAc)2, 2-methylpropanal, O2 | |
| 5 | RuO4 | |
| 6 | Mn(OAc)3, 2 equiv. | 3% |
| 7 | (NH4)2Ce(NO3)6 | |
| 8 | Cu(OAc)2 | |
| 9 | t-BuOK, KMnO4 | |
| 10 | Na2Cr2O7 | |
| 11 | Pd(OAc)2, benzoquinone, LiOAc |
- Reactions performed for 24h at rt in
AcOH, unless otherwise stated; - Complex mixture of oxidation products,
no desired product detected by TLC - Numerous side products including
products of iodination; - Conditions described by Das et al. for
polymer supported cobalt25; - Most starting material recovered
- Reaction performed under standard
conditions described by Sharpless26; - The anion was formed by the method
described previously8, followed by the
addition of KMnO4 and subsequent
stirring at rt for 24h; - Reaction performed under standard
conditions described by Bäckvall.27
| Reagentsa | Ketone | Yield | |
1 |
Co(OAc)3 | 5 | 38%b |
2 |
Co(OAc)3 | 5 | 51%b,c |
3 |
4 eq. Co(OAc)3 | 5 | |
4 |
Co(OAc)3, H2SO4 | 5 | |
5 |
Co(OAc)3, F3CCO2H | 5 | |
6 |
Co(F3CCO2)3 | 5 | |
7 |
Co(OAc)3 | 8 |
- Reactions performed for 24h at
rt in AcOH with two equivalents
of the cobalt species unless
otherwise stated - Numerous side products
incl. products of N-oxidation - One equivalent of Co(OAc)3
was added and the reaction
stirred for 24h, followed by
the addition of a further
equivalent of Co(OAc)3 and
stirring for a further 4h; - Complex mixture of oxidation
products, no desired
product detected by TLC; - Majority of starting material
recovered, no desired
product detected by TLC; - 1 equivalent of additional acid.
A complex mixture of materials resulted from all reactions which made isolation and identification of the products difficult. MS analysis of the mixtures showed signals corresponding to various oxidized products, however it was decided that further analysis was not useful in most of the cases. Of all the reactions, only the cobalt mediated oxidation (entry 2) appeared promising. Although giving just a minor yield of 6, it was noted by MS that the major side reaction appeared to be iodination when periodate was employed, and the loss of material with the use of Oxone was shown to be due to direct reaction of Oxone with 5.28 In order to remove the complications obviously caused by the cooxidant, it was decided to investigate the reaction using stoichiometric quantities of Co(OAc)3, a reagent that can be easily formed by the ozonolysis of an acetic acid solution of Co(AcO)2·4H2O.29 The results of these Co(III) mediated oxidations are summarized in Table 4. It was found that treatment of 5 with 2 equivalents of Co(AcO)3 in acetic acid at room temperature led to a 38% yield of the desired 6 in a very convenient reaction (entry 1). Further study of the reaction showed that the initial addition of 1 equivalent of Co(OAc)3 and stirring for 24h, followed by the addition of a second equivalent and stirring for a further 4h led to an improved 51% yield of 6 (entry 2). Yields could not be improved further due to competing N-oxidation to N-formyl and N-nor derivatives (detected by MS), however the presence of the nitrogen lone pair was shown to be essential as the addition of 1 equivalent of H2SO4 inhibited the reaction (entry 4), and carbamate 830,8 was inert to the reaction conditions (entry 7). These results suggested that the basic tertiary amine was indeed acting as a ligand for the cobalt and directing oxidation to the correct position, however when coordinated, oxidation of this amino ligand was a competing reaction.31 Although allylic oxidation has been observed with Co(III) salts,32,33 the reaction is less favorable than either benzylic34 or aromatic oxidation35 underscoring the importance of coordination of the metal to the teriary amine36.
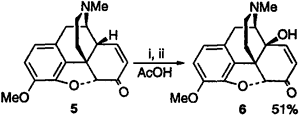
(i) 1 eq. Co(OAc)3, 24h, (ii) 1 eq. Co(OAc)3, 4h
Interestingly, the treatment of 5 with Co(F3CCO2)335 led only to a complex mixture from which no product could be readily identified, clearly showing the dramatic influence of the carboxylate ligands employed. The current work confirms that the direct oxidation of 5 to 6 cannot be performed with the vast majority of common oxidizing agents, however we have shown that this transformation can be conveniently performed by the use of Co(AcO)3 in acetic acid at room temperature. This simple procedure gives comparable yields to the improved MnO2 oxidation17 but, importantly, offers a truly practical method for the direct oxidation of 5 to 6.
In summary, we have developed a practical 1 step synthesis of 14-hydroxycodeinone from codeinone by the use of Co(OAc)3 in 51% yield without the need for chromatography.
Experimental
General Procedures
The spectral (NMR, MS, IR) and other physical data (mp) for 6 agreed with literature values and it was shown to be identical to an authentic sample.10 Peracid oxidations were performed as described in the example below for dimethylacetic acid with the differences noted in Tables 1 and 2. Reactions employing stoichiometric metal oxidants were performed as described below for Mn(OAc)3, and reactions employing catalytic metal oxidants as described for Co(OAc)2/NaIO4 below, with the differences noted in Table 3. Stoichiometric Co(III) mediated oxidations were performed as described for Co(OAc)3 (procedure D) below, with the differences noted in Table 5. The best result obtained is presented in Procedure D, Method 2. For reactions that greatly differ from the examples below, reference to the literature oxidation procedure followed is given in the relevant Table. All reactions were performed under an atmosphere of argon, and all solutions were dried with Na2SO4 and evaporated to dryness on a rotary evaporator unless otherwise noted. H2O2 was a 30 wt. % solution in water, H2SO4 was a 10% solution in water by volume.
- Oxidation Of 5 With Dimethylperacetic Acid
Hydrogen peroxide (0.2 mL, 1.95 mmol) was added slowly to a solution of 5 (500 mg, 1.7 mmol) and H2SO4 (1 mL, 1.9 mmol) in dimethylacetic acid (5 mL). After stirring for 24h, the mixture was diluted with water (30 mL), excess oxidant was destroyed with NaHSO3, and the solution basified with NaHCO3. The products were extracted into CHCl3 (3 x 50 mL), washed with brine (100 mL), and dried. After removal of the solvent, 6 was purified by recrystallization from EtOH with a trace of CHCl3 (195 mg, 37%). The 6 obtained gave spectral data identical with an authentic sample.
- Oxidation Of 5 With Stoichiometric Manganese (III) Acetate
Mn(OAc)3 (450 mg, 1.7 mmol) was added to a solution of 5 (250 mg, 0.84 mmol) in acetic acid (2 mL) and the mixture stirred for 24h. The reaction was worked up as in procedure A to give the crude products. Column chromatography (Silica, EtOAc, NH4OH (0.5%)) gave 6 (8 mg, 3%).
- Oxidation Of 5 With Catalytic Cobalt (II) Acetate And Sodium Periodate
NaIO4 (95 mg, 0.44 mmol) was added to a solution of Co(OAc)2·4H2O (5 mg, 0.02 mmol) and 5 (130 mg, 0.44 mmol) in acetic acid (2 mL), and the mixture stirred for 24h. The reaction was worked up as in procedure A to give a mixture of products. Column chromatography (Silica, EtOAc, NH4OH (0.5%)) gave 6 (16 mg, 12%). MS analysis of the balance of material showed iodinated products (m/z 423; 5+126).
- Oxidation Of 5 With Cobalt (III) Acetate
- Method 1
- Co(OAc)3 (2.7 mmol), prepared as previously reported,29 was added to a solution of 5 (400 mg, 1.35 mmol) in acetic acid (4 mL), and the mixture stirred for 24h. The reaction was worked up as in procedure A and 6 purified by recrystallization from EtOH with a trace of CHCl3 (160 mg, 38%).
- Method 2
- Co(OAc)3 (1.35 mmol), prepared as previously reported,29 was added to a solution of 5 (400 mg, 1.35 mmol) in acetic acid (4 mL). After stirring for 24h, additional Co(OAc)3 (1.35 mmol) was added and the solution stirred for a further 4h. The reaction was worked up as in procedure A and 6 purified by recrystallization from EtOH with a trace of CHCl3 (215 mg, 51%).