Something that was drawn to my attention regarding the content of this document
was that there is no warnings about the materials discussed with regards to
the dangers and effects of the materials produced, and not enough about the
warnings about some of the chemicals used (in particular, shellite/naptha).
ABOUT DMT
DMT is widely regarded as the most potent psychedelic drug known. As has been
said by someone else, the term 'mind blowing' could have easily been invented
to describe this drug. It is not a 'high', or a party drug or any such
thing.
To briefly sum up some of the sorts of nasty experiences that a person can
have under the influence, it ranges from an experience of dying, being torn
apart by vicious animals, being probed and having strange implants installed
by cold uncaring aliens, extreme experiences of humiliation...
Now, after reading that list, one might wonder why a person would be interested
in using this drug at all... Well, basically, because things completely off
the other end of the scale can also happen during the experience. Contact with
happy elves or clown-like entities, exchanges with entities who wish to teach
you something, healing experiences, positive contacts with aliens.
The way it affects people is extremely idiosyncratic, but I can say one thing
that is common to all experiences - it is always humbling, the awesome potency
of this drug should not be underestimated.
MONOAMINEOXIDASE INHIBITORS (MAOI's)
The use of monoamineoxidase inhibitors, such as syrian rue, banisteriopsis
caapi or synthetics such as moclobemide, which is a common way to activate oral
doses of DMT, should be done with great caution. There are elements in most
people's diets and drug intakes which, when combined with the use of MAOIs can
lead to a serious medical emergency if proper precautions are not taken prior
to ingesting MAOIs.
Monoamineoxidase inhibitors (MAOIs) should not be taken
in if any stimulants (caffeine, amphetamines, cathinone, methcathinone), or
SSRI antidepressants (prozac, citalopram, zoloft etc) have been taken less
than 48 hours beforehand, the same goes for beer and wines, and there are foods
which contain proteins which are dangerous if consumed within the same period
of time, these are generally aged protein products (wurst, jerky, salami) and
yeast-based products, such as yeast extracts, wines, beers and other brewed
or fermented materials. Bread is not so bad, since the yeast is still fairly
fresh when it is eaten, but at least 12 hours should be witheld to avoid problems.
The main issues are aged proteins, which covers brewed alcohols, aged protein
foods, and SSRI's and amphetamines are contraindicated, though the info on the
dangers regarding MAOIs mainly applies to synthetic MAOIs (eg moclobemide).
I have been told that syrian rue and harmala do not cause significant risk with
amphetamines and fresh yeast products, however they do potentiate amphetamines.
It is best to avoid them if you are unsure or have not done small scale tests
to ensure it is safe...
In most cases, one will only get a nasty headache, but it is best to avoid
any of these things with MAOIs, recently a teenager made CBS news from mixing
a MAOI with something of this nature (and of course the news people, not really
interested in the propagation of safety information, didn't say what it was
that caused the problem, however it would have been one of the above, as you
can see it covers a lot of common foods and drugs).
What happens is the MAOIs prevent the action of the monoamineoxidase by preventing
it from being synthesised, which stops these enzymes from breaking down chemicals
in the brain and blood to prevent their levels building up too high. The natural
cycle in plasma is of precursors, such as amino acids, being synthesised into
neurotransmitters by enzymes, and these neurotransmitters only have a limited
useful life in the blood as free radicals and other agents can break them and
cause toxicity problems (which is the suspected but not officially recognised
cause of people going bezerk on prozac), and so they must be removed after a
certain amount of time. Also a number of chemicals cause dramatic changes in
physiology, such as stimulants, and the protein tyramine, found in aged proteins,
which is a product of yeast I believe, and the monoamineoxidase enzyme is responsible
for protecting us from this toxicity.
When an incident occurs with MAOIs, a mild incident will involve a headache,
but if too much poisons are in the blood when the MAOI is ingested, they remain
active, and what is called a 'hypertensive crisis' occurs. This means severe
headache, high blood pressure and heart rate. If the
blood pressure gets too high, a stroke can occur, and beyond that, death from
major brain haemorrhage.
SAFETY WITH CHEMICALS
In this document are described the use of numerous chemicals, the ones of most
concern are the Non polar solvents (turpentine and naptha/shellite) and sodium
hydroxide.
I recommend the use of solvent resistant gloves whenever contact may be had
with the solvents. One should be able to find nitrile gloves in the supermarket,
they are marketed as 'non allergy', some people are allergic to latex apparently
(personally I hate latex anyway, it makes my eyes red), there is also neoprene
gloves available, but these will be easily distinguished by being thicker and
'insulating' will be mentioned on the packaging somewhere. Nitrile is the best
to use as it is highly resistant to turpentine and shellite.
Turpentine is not so bad, and being such a high boiling material,
it's vapours are unlikely to develop to an extent to cause breathing problems.
However, shellite is very volatile, more volatile than petrol, and probably
more explosive than petrol.
No sources of flame or sparks should be present when working
with all nonpolar solvents, and one should take care to avoid inhaling the vapours
as much as possible. Always work in an area with good ventilation when working
with shellite.
I have personally been burned by an explosion with shellite, it was only second-degree
(the skin was fully damaged, but it hadn't gotten down to the flesh). I was
in pain for three days, constantly spraying it with xylocaine spray to stop
it from being so painful I was unable to fall asleep. It also put carbon all
over the space that it exploded in. This was from inadequate ventilation and
a spark from moving a pot around on an electric hotplate while blowing the vapours
out of the pot. The vapours of shellite/naptha are heavier than air, so they
will fall downwards, so it is particularly crucial that there is nothing incendiary
below any amount of such materials at close to it's boiling point. The vapours
will also flash back to the source of vapour for quite a distance, at least
two metres clearance should be given before anything with a slight risk (including
a mobile phone) of causing a spark should be permitted nearby. It is also for
this reason that, unlike numerous other guides, I don't
recommend heating the solvent when extracting. Besides everything else,
experience tells me that one does not need to increase the solvation power of
300ml of naptha to get everything desirable out of an expected 1-3g yield. The
only time when it is sensible to heat naptha is when recrystallising, and this
can be done in open space using boiling water from a jug.
Sodium hydroxide (NaOH) is also very dangerous, anyone who has seen the movie
'Fight Club' would recall the scene where Tyler Durden puts a pile of this material
onto the protagonist's hands. When it comes into contact with water it reacts
with a very strong exothermic reaction (it generates heat), and for this reason
the NaOH should always be added to water and not the
other way around as this ensures that the heat is absorbed by the water,
rather than the heat inducing the hydroxide to react violently with the humidity
in the air, and it can in fact explode, spraying highly reactive material everywhere,
and when this gets on the skin or in the eyes, it burns severely.
It is recommended when working with sodium hydroxide
to wear protective goggles in case it splashes in the face, it can destroy your
vision .
Acknowledgements
I would like to acknowledge the contributions of various people who have helped
me gather the information I have assembled in this document. Thankyou to Yjanni,
Mescalito Ted, Yoda, Quantum Tantra (for her wonderful guide), Meme (for the recrystallisation
information and for sharing my passion for improving techniques and administration),
Carpedmt (for highlighting the fact that extractions do not need take a long time,
and for introducing me to acacia obtusifolia), Silicon7 (for his knowledge of
proper chemistry procedure and help with materials), and his cat Moofie who's
presence in the lab greatly amused me, Rhodium (who's excellent chemistry knowledge
and great information resource http://www.rhodium.ws
and the hive http://www.the-hive.ws), Gastro
(who provided my first few quantities of acacia extract many years ago), Mulga
(for his very informative and useful site), Mesqualero (for moral support and
for inspiring me to create a more clear format for the instructions and notes),
Andrew (for his report), Eccles for his excellent pKa info on DMT, Glorfindel
(for pointing out the need for pH 12.5 or higher for polar washing which, sadly
are a bad idea as it turns out), Gumby, for emphasising the safety factors, Bluetonguedragon
for his encouragement, Ion for his excellent explanation of eliminating tannins
and dry extraction techniques (which I don't explain here), all the people who
have emailed, pm'ed and instant messaged me with praise for this, and anyone else
who I have gathered information regarding this process who I might have forgotten.
I have written this guide to specifically cover the process of extracting alkaloids
from the Acacia Obtusifolia tree's bark, which is the most consistently yielding
of the acacia species with regards to it's yield of psychedelic tryptamines,
primarily N,N Dimethyltryptamine. It is equally applicable to mimosa hostilis,
and is likely to be useful for any other DMT source contained in the bark of
a plant (eg, desmanthus illinoensis). Chlorophyll laden sources, such as phalaris
and psychotria viridis et al have not been studied by the author, however at
some point in the future the special considerations for these leaf materials
will be added.
One thing that I have learned in recent months about the process of extracting
alkaloids is that the long periods of time suggested in older guides are unnecessary,
this suspicion started to come to me when I witnessed the acid/base extraction
of alkaloids from an alkaloid synthesis and saw that the alkaloids can in fact
cross phases in a matter of minutes, not hours or days.
Taking care to ensure certain pH levels are used also eases and improves the
process, specifically in the acid phase the pH is better if it is about 4, and
in the extraction less emulsion problems occur when working with a pH above
12.5 (sodium hydroxide does not raise the pH above 13.5 when diluted 5g/100ml,
so the concern about 'pH spikes' is unnecessary so long as the base is diluted).
When washing and extracting, use gentle agitation rather than violent, swirl,
stir or turn it end over end gentlyrather than shaking it. Water washing is
less of a problem in this respect, usually by that stage the majority of emulsion
causing chemicals have been removed.
Something else which I have heard from others, which I have now verified, is
the value of doing washes. Previously washing was little mentioned, and only
the wash of the acidic extract was suggested (and again, a much longer than
necessary time stated), when in fact the best results seem to be from doing
a non polar wash of the acidic extract, and using a fairly low pH (like 8-9)
and no salt, for washing the non polar extract (the best proceedure seems to
be using sodium bicarbonate as it reacts with sodium hydroxide to produce sodium
carbonate, which more readly comes free of the nonpolar fraction). Washes ensure
the resulting product is as free from contaminants of a non-alkaloidal nature
as possible, in particular residuals of the base being used. Again, gentle agitation
is completely sufficient for extractions and washes, and in the case of water
washes, violent agitation will not allieviate suspended droplets of the base,
the bubbles of the wash water, being alkaline, will wash some out, but will
also leave the suspended bubbles of base in the nonpolar solvent, though by
this stage in the process it doesn't matter as much as it should be quite clean
already.
Finally, the best methods for recrystallising the product seem to be coalescing
for me as well, I will talk about this at the end, the purpose is not, properly
speaking to make pretty rocks, though if large crystals are produced they have
a longer shelf life due to lesser surface area per volume, but instead, the
purpose of recrystallising is to eliminate the contaminants in the solvent commonly
used (naptha), as well as reducing other contaminants. This requires slow cooling
or progressive cool/warm steps.
This guide will not attempt to cover the extraction processes for other plant
materials, however it is intended to be comprehensive and to the point, and
should apply more or less to other materials.
I will also focus on the equipment which will ensure the best results with
the least outlay without any compromise of efficacy of the process.
This guide is specifically targeted at the acacia obtusifolia, I cannot really
enlarge upon other information already available through libraries and on the
internet, but I intend to replace this section with a comprehensive guide to
finding them based on a series of expeditions to likely locations and lots of
photographs, and, of utmost importance, in plain english.
One of the problems with the botanical information regarding these plants is
that they never go into much detail about the bark structures. When walking
through a well developed forest, quite often the tree is so far above you that
you can't see a thing of what they describe in the botanical description. As
soon as possible, a photographic expedition will be made to bring back good
photographs of the plant in it's mature natural state. The full extent of the
obtusifolia's habitat is not fully known, it may well be more common than is
currently thought.
The guide should be applicable to other sources of DMT, as the method described
is very thorough.
Other, more advanced chemistry equipment can be used for this process, however
my aim is to only talk about what I believe to be the minimum standard to ensure
the best possible results.
Large Cooking Pot - for beginners it is wise to start small,
2-4L soup pots are good for this. Do NOT use aluminium
pots for this stage, even organic acids will take up some of the aluminium
oxide on the coating and the aluminium will react with strong acids to produce
aluminium salts (eg aluminium chloride) and this is not something that one
wants to need to get rid of, let alone ingest.
Glass baster - this is a large pipette used to suck up
oils from fowl being roasted in the oven and coating them so they get nice
and golden crispy on top. A baster has a long glass tube and a rubber squishy
thing at the end and it works like a dropper. Generally they can hold about
30-40ml. Glass is essential because these devices can be used to separate
immiscible phases. These devices are available at most kitchen shops. I just
want to emphasise again - make sure it is glass - some of the solvents used
will attack plastics (especially nylon, which is the common plastic baster
material because of it's high melting point) and metal is completely useless
for this purpose. Also it is good if the device has a tip on it which is very
narrow for half a centimetre or so, this improves this device's ability to
precisely separate fractions and how well it holds the fluid when removed
when held mid-air.
pH meter - In my opinion it is worth the money to get
a good pH meter. These can be purchased from hydroponics shops and aquarium
shops, they are commonly called 'pH Pens' or something similar. When getting
a pH meter be sure to get an ample container of pH 7 'buffer' solution for
calibrating it, and do this at least once a month. pH papers can be used,
however the extract has a very dark reddish colour and stains the papers.
The only type of paper test which is suitable is the type with four different
tests on each strip, and the reason why I suggest using an electronic tester
is that dollar for dollar, it is hard to see what the effective advantage
is, for the cost of 400 strips one can have an electronic one, and the electronic
testers are accurate (if calibrated correctly) to about ±0.05 pH units,
though this is not critical, using precisely measured pH levels every time
is helpful in generating consistent results as the exact pH levels influence
solubilities of various constituents of the plant material, and the best policy
is to try and pick a pH which dissolves what you want and as little of what
you don't want. Bear in mind that every extraction requires about 10 individual
pH tests, thus for the same money as you can do 400 extractions with paper
tests, you can do well over 1000 with a pen.
Large Jar - a jar like the ones that cranberry juice is
sold in is perfectly suitable for the task, though a 2L canning jar can serve
quite well also. If a canning jar is used, it should be tested with turpentine
to ensure that the rubber used on the seal does not get damaged by it. The
juice bottles also have the advantage of being able to rapidly vent, the clips
on canning jars require much more time to open when venting. Shellite is safe
to use with most common HDPE plastic bottles, but these bottles are unsuitable
for use with aromatic solvents such as turpentine, xylene or toluene, which
I suggest using. An empty turpentine bottle is suitable also. This is the
container that the extractions and washes will be done in, so it needs to
be as large as the quantities of material you work with, and the opening big
enough to permit the use of the baster.
Colander - to catch bulky fibrous material from the acidic
extract.
Strainer - This will catch the larger particles.
Fine Strainer/Tea Strainer - this will catch medium sized
particles, which will block the coffee filters.
Cotton, Acrylic or Polyester felt - common pillow stuffing
can be used here, so long as this is done before doing the Non polar washes.
This material catches a lot of fine particles, it functions as a
'depth filter', which is a thick filter which is used to remove the majority
of large and medium sized gunk before using filter papers. This kind of filter
will also be helpful with the gunky gooey materials that are sometimes present.
Coffee Filters - to filter finer particles out of the solution.
Particles of bark will make seeing the line between phases in the Non polar
wash difficult to see. The coffee filters can be good for removing hairs and
other junk when recrystallising as well.
Pyrex Measuring Jug - this is helpful with filtration and
removing extract from broad containers (such as cooking pots) as well as measuring
solvents for extraction and washing.
Drying Dish - The dish should be deep enough to accommodate
the amount of solvent used. Ceramic plates can be used too, ones with gentle
curving edges and plain white glaze are quite good for being easy to scrape,
though they can only have about 100ml in them.Several such plates would be
necessary for an extraction - this can be a good thing for seeing what each
consecutive pass gets out.
Scraper Blades - These are essential for scraping up the
final product, the best ones are the single sided ones used for removing stickers
from glass. They have the advantage of being easy to handle and able to bend
slightly for getting around corners in drying dishes.
Acid - I recommend tartaric acid,
which can be bought from supermarkets from the cakes and lollies section.
Other acids can be used, but hydrochloric acid, in particular, should only
be used with fully intact enamel pots or with pyrex pots. Tartaric acid is
handy because it only takes a teaspoon to turn 10L of water to around pH 3-4.
Citric, acetic, and ascorbic acids can be used too, though beware of acetic
acid if DCM is used in nonpolar washing as the acetate salts of DMT are fairly
soluble in DCM
Base - Sodium Hydroxide (NaOH -
often sold as drain cleaner, but be sure that the drain cleaner you get says
'99% sodium hydroxide', it is also known as 'caustic soda' and in the US the
brand Red Devil Lye is allegedly good - in fact anything called 'lye' is a
base, but traditionally it was potassium hydroxide), Potassium Hydroxide (KOH,
sold at hydroponic shops as pH up, and used to make liquid soaps). NaOH and
KOH are both strong bases and should be dissolved 1:10 at least before using
(premixed solutions from the hydro shop are fine as is). Some suggest using
ammonia, but domestic sources of it are not as clean as NaOH, thus I do not
recommend it. Always add sodium hydroxide and potassium hydroxide
to water, and not the other way around. When working with strongly alkaline
solutions it is recommended to wear safety glasses and gloves.Read
my warning for more information
Non polar Wash Solvent - Turpentine
(paint thinner, available anywhere, dissolves oil paints), toluene (commonly
used as an octane booster, also used to dissolve vinyl glues, for laying vinyl
flooring), and xylene (available in many hardware stores, I have heard of
it being used to remove the resins that are used to coat tiles - presumably
urethanes). The aromatic solvent is used in this technique because it dissolves
a lot of stuff, in the defatting stage, where a solvent which clears a lot
of gunk is useful. I recommend turpentine for the simple reason it is much
easier to get, much cheaper, does the job just as well, and is less
toxic.
Extraction Solvent - There is basically
three choices for this, and which one used determines how one must do the
separations. Ethyl ether (from automotive stores as starter spray) and methylene
chloride (also known as dichloromethane, or DCM) both share the property of
having a very low boiling point (around 40°C) and consequently, when using
them at room temperature, using a turkey baster, sucking up these solvents
will be an exercise in frustration. The solvents evolve a lot of gas, often
at higher than ambient air pressure, so they pump themselves straight back
out. Thus if a turkey baster is used and the Non polar is one of these, one
must remove the polar phase, as this will not cause the same problem. With
DCM this means pulling the top layer (DCM sinks) and ether, pulling the bottom
layer. A separatory funnel makes the use of DCM much more enjoyable, since
the tap is at the bottom and that's where the DCM ends up too. DCM also absorbs
a lot of the desirables very quickly. Naptha (aka shellite) is readily available,
and is easy to work with using a baster. Be very careful with shellite and ether, they are
highly volatile and explosive.Read my warning
for more information. Dichloromethane can damage the lungs, care should be
taken when using it too.
Recrystallising Dish - From limited
experience, it seems that the best container for this is a dish which has
a curved inner surface (like a noodle bowl), and plastic wrap is used to prevent
the solvent from rapidly escaping. Scraper blades will probably need to be
bent into a curve to scrape the edges properly, though a scalpel might serve
better for removing the crystals. A double layer of plastic wrap is good for
ensuring that it doesn't stretch down too much when it cools (the gas trapped
inside will contract when it cools).
Recrystallising Solvent - Due to
it's higher boiling point than any other available, and it's fairly reliable
homogeneity, if possible, the best solvent is isopropyl alcohol (IPA). Naptha
will also work, though the lower the boiling point of the solvent, the more
it will evaporate away while handling, which can be a pain. Acetone can probably
be used, as can methanol and ethanol. Ethanol seems to need very little to
dissolve the material, which can be problematic with very small quantities
(a gram or less). Slightly polar solvents are good in this step as tannins
dissolve quite well in them, if they get to the final product. Acetone and
ethanol have not been tested as far as I know, but I do know that acetone
dissolves DMT freebase quite well
Dropper Bottle - This is used to
transfer the hot recrystallisation solvent into the recrystallisation dish
(specifically, the dropper)
Nitrile Gloves - most domestic glove
manufacturers now sell a special kind of glove for people who are allergic
to latex (IMHO latex is nasty anyway), there are two types, neoprene and nitrile,
the former are thicker and are particularly good for handling hot liquids,
the latter are good for handling Non polar solvents, particularly aromatics
which will very rapidly decompose latex. I have come to the conclusion that
it is crazy to work with these materials without using good protective gloves.
In particular when agitating washes and extractions involving highly volatile
solvents (ether, dcm, naptha) this permits venting of jars without exposure
to the solvents.
Safety Glasses - This is recommended for any stage involving
the use of sodium hydroxide, and to a lesser degree, non polar solvents, as
protection against damage to the eyes that can occur. In particular, sodium
and potassium hydroxide can burn the eyes and even blind you if they damage
the eyes enough.
The bark must be prepared for extraction, a simple but effective method that
can be used is to simply pull strips of fibres off using fingernails (or assisted
with a pocket knife) so that thin pieces are removed - this must be done when
it is still fresh. The bark can be cut up into small pieces and put through
a blender or coffee grinder, this is best done when dry, though cutting it up
when dry can be difficult, it would be best done when still fresh (it is best
to ensure cutting goes from the inside of the bark to the outside because the
inner fibres are long and resistant to being cut). If bulk quantities are being
worked with (like 10kg or more) a mulcher can be very effective. A small garden
shredder is very helpful in any case, and for anyone intending to powderise
the material, it needs to be broken into small pieces before putting into a
blender.
The more the bark is munched up the faster and more thorough first two steps
are.
This step uses an acid to make the
tryptamine alkaloids soluble in water - the acids ionically bond weakly
to the amine tail of the tryptamines and render them soluble in water
like any other salt.
This permits other elements in the solution which are less soluble
in water and sparingly soluble in water to be removed in the next step.
Prepare a solution of water with the pH adjusted
to 3-4 with the acid you are using.
Add to the plant material to the solution, either
soak (a couple of hours for powdered material, overnight or longer for
less finely shredded material) or simmer for 1 hour.
Pour off the liquid and repeat two more times
with more acidified water.
Combine the extractions and reduce
on gentle heat to a workable quantity (about 1L)
This step uses the fact that the alkaloids are now well dissolved in
water to permit the use of an aromatic solvent (which would readily
dissolve the alkaloids in freebase form but not as the salt) to thoroughly
strip anything from the solution which will contaminate the product
at the end of the process.
There is significant likelihood that the bark contains small amounts
of histamines, which will be removed by this as well; in susceptible
persons, histamines will cause the throat to constrict and make breathing
difficult when smoking the product, which is an unpleasant side effect
that is best avoided.
Aromatic solvents are suggested in this step because they dissolve more
than any other Non polar solvent, and thus clean the product more than
ether, chlorinated solvents or naptha/shellite.
Pour the acidic extract into the separating container
(separatory funnel or jar).
About 100ml
of the aromatic solvent (toluene or xylene, or at a pinch, 200ml of naptha)
is added and the jar is gently agitated (swirling, stirring, end-over-end)
Aromatic solvents are used in this step because they
have high solvation power, and will remove anything that might
contaminate the final product.
Adjust the volumes of solvent upwards in the case of lesser solvents
(such as naptha) to ensure it pulls as much of the gak as possible,
also use more solvents if one is working with a larger quantity of material
Separate
the aqueous phase from the aromatic solvent phase.
Repeat the wash with the aromatic solvent two more
times.
The reason for doing more non polar washes is that the primary contaminants
in the product is usually from not doing sufficient washes at this stage
Do a final wash using the solvent that will be used
for extraction to remove any excess aromatics suspended in the
solution.
This is done to eliminate the majority of the high boiling aromatic solvent
from the solution so when it is extracted the high boiling component
of the solvent is reduced. If there is materials that boil at a significantly
higher temperature than that of the DMT (i.e. much more than 120°C)
the DMT will not crystallise or dry properly, as it will boil before
the solvent it is mixed with. This is known as 'oiling out' and is best
avoided if possible.
In this step the pH of the now washed extract solution is raised to 13,
which will drive the alkaloids out of the water by binding up the acids
that caused the alkaloids to be water soluble.
Mix up a solution of the base - for sodium hydroxide,
5 teaspoons to 250ml is sufficiently dilute.
If one is concerned about pH spikes from introducing a
solution this concentrated, twice as much water can be used. The
pH of the solution made via the instructions to the left is
around 13.5 or so, and really it isn't too strong. The main thing is
adding it slowly while stirring.
Add
the alkaline solution slowly to the acidic extract, stir well
while adding the base.
There are many ways to do this, some people use a
baster to transfer the solution, and use it to stir and pump the
solution around, it can also be added slowly by pouring it in
while stirring with a skewer.
I don't recommend basifying in one container and then transferring
to another. The DMT can stick to a lot of plastic and glass surfaces,
and ideally one wants to get as much of it as possible.
Making the pH to 13 drives the tryptamines back into freebase form, in
which they are insoluble in water. They are, however soluble in Non
polar solvents, such as the ones described as suitable extraction solvents
(ethyl ether, DCM and shellite/naptha). This property permits the separation
of the freebase from the basified aqueous extract solution. Using a
pH this high also eliminates emulsions.
Add a quantity (100ml is a good amount for an
extraction with an expected yield under 3g) of the extraction solvent
to the basified solution.
The amount of solvent to use depends a lot on how much
you expect to get from the plant material.
As a rule of thumb, 100ml of naptha/shellite will dissolve about 1g
of DMT, less is needed for dichloromethane and ether, though the amounts
must be kept practical for the separation method being used.
Also note that using an excess of solvent may result in more being
extracted than desired. This is particularly the case with naptha.
Close the
container gently agitate, periodically vent to release pressure
Some guides recommend heating the extraction solution to increase how
much it draws across when doing this, I must stress that this will dramatically
increase the amount of pressure that will be generated by the agitation,
having almost lost my vision from not venting a jar of hot shellite
and having the jar pop and spray everywhere, I don't recommend heating
the solution at all.
Some guides say 'keep the base cool and the solvent warm' - this is
not physically possible, the heat in the total solution will very rapidly
equalise, especially if it is shaken vigorously.
Separate the nonpolar solvent from the basified
solution.
Because the pH is
high, at around 13, and because the solution has been thoroughly washed,
it will not form stubborn emulsions, the differentiation of the layers
usually is almost perfect within 5 minutes.
It is not neccessary to wait long - 5 minutes of gentle agitation and
10 minutes of settling is usually long enough for each pass
This step is done to eliminate any residual polar soluble contaminants
present in the Non polar extraction. The main concern is the base itself
(which burns hot and makes the product harsh), though there is also
problems involving contamination from the aqueous fraction also, plant
substances etc.
Generally speaking the stuff washed in this step will not
prevent crystallisation if left in, but it should not be left in
because it is just yucky.
Add an excess of water adjusted to a pH of at least 8.5 to the non polar
extract solution.
Agitate the mixture gently for about 5 minutes,
let it settle before proceeding
Gentle agitation is essential to ensure that no microbubbles of alkaline
wash water remain in the nonpolar extract solution. If violent agitation
is used, one should use plain tap water, but I don't recommend violent
agitation, basically it is unneccessary
Remove the water from the nonpolar extract,
and repeat this wash two more times.
Repeating the wash three
times will ensure that none of the base or other polar soluble undesirables
remain in the solution. This step is critical for removing residual hydroxides
and salts of the acid and base being used. If this step is not done properly,
the hydroxides will react with the freebase and destroy them, not to mention
burning the throat.
Next, put the combined Non polar extracts onto a drying dish, something that
will permit the easy scraping of the materials.
Using an electric fan to blow the drying dish vigorously is a good idea, it
dramatically increases the rate at which the solvent evaporates off.
If the proper equipment were available, a vacuum desiccators would be even
faster, and would permit recovery of the solvent, though this is not accessible
to most people who wish to do this process. The shellite evaporates very quickly
anyway, and ether and DCM are even faster.
Although it probably helps with getting all of the extract out of the dish,
it is not absolutely necessary to bring it fully to crystallisation at this
stage. This will take at least 12 hours to fully happen under a fan. The extract
is easier to remove from the drying dish if it is fully dry as it has a much
harder consistency and comes free of the glass much more readily.
This step is done to refine the product further, and indeed can fully
purify the extract to translucent crystals. The longer this stage is
done, the larger the crystals can become.
When recrystallising, the idea is to use a solvent which dissolves undesirable
elements better than the desirable element, hence with study, the best
possible solvent or mixture of solvents can be developed to clean the
product most effectively.
This step eliminates contaminants in the product which will make it
harder to smoke.
Heat the recrystallisation solvent until it starts to boil by sitting
in a container over water heated in a jug.
A good container to put the
recrystallisation solvent in is one which has a dropper in it, as you
can simply let it sit in the water until it gets to the same temperature
as the water, and then you can immediately drop the solvent onto the freebase.
Place the scraped up product from the previous
step into the recrystallisation container and allow the container to
get nice and warm.
Be careful here not to let the freebase get so hot
that it boils, as this will cause a loss of product
Slowly add the hot solvent to the warmed recrystallisation
container, stirring and agitating until all of the product is *just* dissolved.
The idea here is that the minimum
amount of solvent, at the highest temperature possible (at which it will
dissolve the most possible product for the volume), which, when cooled
will not dissolve the product very well at all, and thus it will precipitate
as crystals.
Seal the recrystallisation container.
A jar can be used for recrystallisation - when the
product crystallises hard as it does in this process, it can be scraped
out using a scalpel or a piece of wire.
If using a beaker or bowl, a double layer of plastic wrap is
recommended, and fixed in place using a rubber band.
Sealing the container simply prevents the crystallisation of
water when the container is put in the freezer.
Let the container cool to room temperature.
Alternate between the fridge and outside the fridge every hour or so
for as long as you like.
The colder temperature causes crystals to precipitate,
and then warming them again dissolves the smaller crystals more
rapidly than the larger crystals.
When done for long enough, this favours the formation of large homogenous
(containing only one substance) crystals. In the case of acacia obtusifolia,
there is only DMT and NMT. The NMT forms an amorphous yellow resin,
and with the correct choice of recrystallisation solvent, can be selectively
removed, leaving behind only the DMT, which is a translucent waxy crystalline
substance.
The DMT, in any case, (and I think this applies to 5 methoxy DMT) is
a clear crystal, the yellow or brown or red colour common in extracts
is not from the DMT.
It improves the resulting size of the crystals if the initial room
temperature/refrigerator stage is done for a lot longer, this causes
the crystal cores to trap less contaminants.
Then alternate between the freezer and the fridge for
another period of time.
While still at freezer temperature, discard the
solvent (it has very little in it of value).
Because the extract was
just dissolved at close to it's boiling point, at the
temperature in a freezer there is a very insignificant amount of the
desirable alkaloids remaining in solution. With the right choice of
solvent, the DMT can be preferentially precipitated and thus isolated
from the other components in the extract.
Repeat up to two more times
If done more times, the
extract is much cleaner.
Dry the final recrystallisation.
When the extract
is very pure, the product will be very easy to remove from the container.
A scalpel is a good device for this, one with a curved blade.
I have written this extraction procedure in the hope that others who find the
lack of clarity and good chemistry information in most guides offputting, also
this guide exposes a number of myths regarding times required for processes,
in particular the Non polar extraction. The product resulting from following
this guide, assuming one begins with a good source, should be just about as
pure as the product can be made. Also, if a neophyte follows this process, and
they have started with good material (good material is equally important as
the right technique, you can't get gold out of you put garbage in), is guaranteed
to get a good result.
I will be revising this process until it becomes clear that it covers the
topic in a thorough and concise way, it is my hope that this document will ease
the transition from neophyte to journeyman that can be extremely frustrating
and drawn out. Accurate information will ensure that so long as the directions
are followed, good results will come out at the end.
Reports of using this guide will be appended to the end, I encourage anyone
who has had success with this technique to send me their reports so I can add
this to this document to demonstrate the efficacy of the technique, and to encourage
neophytes to keep trying if they do not do as well as they expected.
One final comment, most people will not get as much as they hope for out of
extractions. Over a process of a number of extractions one gets a better idea
of how much to expect, the visual appearance of quantities of freebase of various
weights will take a while to know, as will the amounts of plant material that
produces it, and in some cases, the right times and ways to collect the material
will also influence the results. Do not be disheartened if yields are lower
than you expect, only experience will teach you how much to expect in practice.
Need I say it, all of these reports are from
people who have done these processes where doing so is legal, and all
are anonymously sent to me to add here.
Mimosa Hostilis Extraction (13 April 2003)
Materials; A few 500ml flasks/beakers, a jar, separatory funnels, vacuum
hand pump, glass funnels,
cotton, cheese cloth, evaporation dish, PH papers. I used Mimosa Hostilis
Root Bark, Muriatic acid (HCL), Lab grade Sodium Hydroxide (NaOH), distilled
water, and Naptha (evaporates cleanly).
I started by boiling 325ml of distilled water, then took it out of
the pan and into a beaker where I acidified to a PH of about 3 or 4
using muriatic acid.
Then I poured this acidic water in a jar containing 88.4 grams of finely
powdered mimosa hostilis root bark. Since I used hydrochloric acid I
decided not to add all three acidic water extracts together and reduce
volume because I didn't want to concentrate the acid.
So what I did was each of the 3 extracts on there own. After putting
the acidic water with the root bark I waited 1 hour and poured the liquid/root
bark into a cheese cloth on a beaker, and squeezed all of the water
out. I then took the dry root bark and put it back in the jar with the
same amount of acidic hot water as the first time.
And while waiting another hour for that to finish up I filtered the
liquid from the first extract into a funnel with cotton and a vacuum
pump connected to the flask.
Then I defatted by putting 80ml of xylene and throwing away the xylene/emulsion.
I defatted 2 more times with naptha(80ml).
Then I basified with NaOH, when it was about half way threw I added
some naptha and kept basifying until it turned a slippery black type
colour. Then I separated the naptha with the emulsion and added 80ml
of warm naptha. I separated that naptha/emulsion and added another
80ml of warm naptha. Again I separated and kept the naptha/emulsion.
Now I took the naptha/emulsion and added some PH 8 distilled water
and threw away the water. This got rid of most of the emulsion, I put
the rest in a graduated cylinder to further separate. I washed the naptha
2 more times with PH 8 dH2O.
I evaporated the naptha via fan and dish to reveal .4grams (400mg)of
white fluffy crystals. By this time the second soak of root bark was
ready to be further extracted so I put that through a cheese cloth and
cotton funnel and then put more acidic water with the root bark.
I defatted this time with only naptha 3 times. Basified and extracted
and washed just as before. I evaporated this to find .22grams of crystals.
I extracted the last of the acidified water by filtering, defatting,
basifying, added naptha, washed naptha and evaporated just as before
to get .06grams of crystals.
I did this whole extraction in under a day, and my yield tells me I
really don't have to wait as long as some teks say. My total yield was
.68grams of spice from 86.4g of mimosa root bark. That's .4grams the
first one, .22 the second and .06 the third one.
As you can see the last extract is low but not completely insignificant.
If I were doing an extraction with a different acid like say acetic(vinegar)
acid I would have combined my extracts and boiled down to about half
or 1/4th it's original volume, but since I used HCl I decided it would
be better to do each separately.
Photographs of Mimosa Hostilis Extraction
Dry Mimosa Hostilis Rootbark
Ground mimosa hostilis rootbark
Mimosa hostilis rootbark soaking in acidified water
Filtering the liquid from the rootbark soak
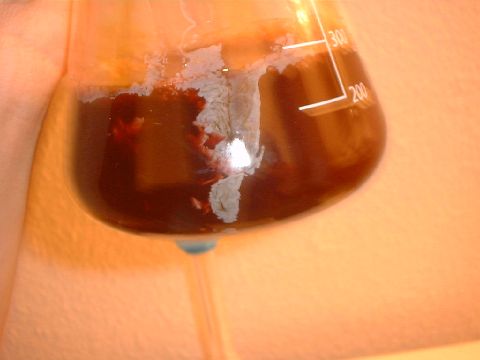
Defatting the acidic extract (see the colour in the naptha layer?)
Basifying (notice how the base immediately makes the solution turn opaque pinkish)
Basifying
Basifying when the solution is fully pink, the pH is between 10 and 12,
Naptha added to basified extract - as it turns black (seen here at the
bottom of the flask) the pH is over 12 - this is how high it needs to
be for proper extraction
First extraction run (see the pink stuff - that's emulsion, wait until
it settles and the margin between the layers is mirror-like)
Water washing (note the dark colour in the lower layer - this is why
you water wash) (also, for this first step, use sodium bicarbonate as
this will eliminate the residual sodium hydroxide, transforming it into
sodium carbonate)
Water washing - see how it gets lighter
Water washing - never stop water washing until there is absolutely no colour in the water being used to wash with
Crystals
Crystals scraped together (note how clean looking they are - this is achieved through proper defatting)
Elfspice: New instruction format implemented - using simple instructions on the
left and notes and clarifications on the right. This makes it a lot easier to
read. The recrystallisation technique was condensed to one step. Thanks to mesqualero
for the idea
14 April 2003
Elfspice: Prettified the two column sections by making all the note columns grey
coloured, added a report, fixed the paragraphing. Thankyou to andrew for the
report.
15 April 2003
Elfspice: modified polar wash directions to specify using water basified to at
least 12.5 after a suggestion from glorfindel from DMT world.
16 April 2003
Elfspice: Added notes about doing further filtration.
19 April 2003
Elfspice: Expanded filtration information, added a 'warning' section (thanks gumby),
added more equipment to the equipment section (strainers, filters and safety
glasses).
27 April 2003
Elfspice: Made minor corrections, refined the notes a little bit, removed the use
of high pH water for the water wash, removed the reference to ammonia as a base
due to it's not being available anywhere near as pure as NaOH, reduced the section
on the acid to only specifying tartaric acid, and mentioned the incompatibility
of hydrochloric acid with any kind of metal.
1 august 2003
Elfspice: added a sequence of photographs showing the all the steps of the process