Abstract
Secondary amines are obtained in moderate-good yields by reduction of crude imines prepared from
N-alkyltriethoxyiminophosphoranes and aldehydes via the aza-Wittig reaction. N-Alkyltriethoxyiminophosphoranes
are synthesized by one-pot azidation of alkyl bromides followed by Staudinger reaction.
Significant interest has developed in recent years in the application of iminophosphoranes for various
synthetically useful transformations, especially those affording C=N bond possessing compounds, which are
usually referred to as aza-Wittig reactions1. Although numerous articles have appeared on the reactions and
synthetic applications of N-alkyl(aryl)triphenyliminophosphoranes2, the preparative utility of analogous
aza-ylides, viz. N-allyltriethoxyiminophosphoranes is almost unexplored3. Recently, we reported the one-pot
preparation of N-alkyltriethoxyiminophosphoranes from alkyl bromides by azidation followed by Staudinger
reaction4 and the application of these compounds for the synthesis of primary amines5. Herein we describe a
route for the synthesis of secondary amines beginning with the parent alkyl bromides and employing a facile
construction of the final carbon skeleton via the aza-Wittig reaction between N-alkyltriethoxyiminophosphoranes
and aldehydes. Secondary amines are synthesized by the variety of methods, the
reduction of imines being a very convenient and explicit route6. Difficulties which may be sometimes
encountered in this approach are due to the properties of some imines which are unstable and therefore not
easily accessible in pure state. We decided to overcome this disadvantage by preparing the respective imines
under mild conditions and use them for reduction without isolation and purification. The synthetic protocol
illustrating the construction of secondary amine skeleton from two simple building blocks - alkyl bromide and
aldehyde is presented in the Scheme.
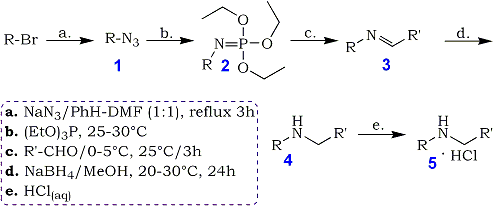
Azidation of alkyl bromides was performed for 3 hours at the reflux temperature of the solvent
system, using a 100% excess of sodium azide. Benzene solution of crude alkyl azides 1 afforded
N-alkyl- triethoxyiminophosphoranes 2 when treated with triethyl phosphite at 25-30°C.
Isolation and purification of these aza-Wittig reagents was neither necessary nor recommended. Neat 2 are
extremely sensitive to traces of moisture and hydrolyze easily to diethyl N-alkylphosphoramidates which may
then contaminate the target compounds. Benzene solution of 2 reacted easily with aliphatic and aromatic
aldehydes to give the imines 3. If one followed the course of this reaction by 31P-NMR, the practically
complete conversion of 2 to 3 was observed after 3 hours at room temperature. Due to the exothermic
nature of this reaction, all reactions were started at 0-5°C and then run at 20-25°C.
Table 1.
Preparation of secondary amine hydrochlorides 5
| Entry | R | R' | Yield* | mp (solvent) |
| 1 |
PhCH2CH2 | Ph | 77% | 259.5-261°C (95% EtOH) |
| 2 |
C6H13 | Ph | 80% | 214-215°C (acetone) |
| 3 |
CH2=CH-CH2 | Ph | 85% | 138-139°C |
| 4 |
PhCH2CH2 | i-Pr | 75.5% | 239-240°C |
| 5 |
C6H13 | i-Pr | 62% |
236-238°C (dec.) |
| 6 |
PhCH2CH2 | Pr | 40% |
215-218°C (dec.) |
| 7 |
C6H13 | Pr | 32% |
248-250°C (dec.) |
| 8 |
sec-Bu | Pr | 19% | 196-197°C |
* Overall yield of crude 5 calculated on alkyl bromide
The crude imines 3 were not isolated but immediately reduced with sodium borohydride in methanol to
form the target secondary amines 4. The amines were purified by steam distillation and characterized as
corresponding hydrochlorides 5. This methodology can be used to prepare a number of secondary amines as
shown in Table 1. The range of substrates in Table 1 demonstrates the clear utility of this synthetic
procedure in preparing a variety of secondary amines in moderate to good yields. Best results were obtained
when benzaldehyde and primary alkyl bromides were used as starting materials. The overall yields of 5 were
inevitably lower (entry 6 and 7) with easily enolizable butyraldehyde as carbonyl substrate, possibly due to
base-promoted (by strongly basic 2) aldol condensation as a side reaction.
Although secondary alkyl bromides could be easily converted into the corresponding iminophosphoranes 24,
the aza-Wittig reactions of the latter were totally ineffective (entry 8).
Experimental
General
Unless otherwise stated, all solvents and reagents were purchased from commercial suppliers and used without
further purification. Aldehydes were freshly distilled before use. Melting points were determined in open capillaries
and are uncorrected. All new compounds gave satisfactory elemental analyses data. Infrared spectra were measured
using a Specord M80 (C. Zeiss) instrument, and NMR spectra were recorded at 80 MHz with a Tesla BS 587FT
spectrometer. All amine hydrochlorides exhibited IR and NMR spectra in accord with the expected structures.
Preparation of Secondary Amine Hydrochlorides 5; General Procedure:
A suspension of finely powdered sodium azide (6.5 g, 0.1 mol) in the mixture of alkyl bromide (0.05 mol),
benzene (15 mL), and dimethylformamide (15 mL) was refluxed gently with stirring for 3 h. In the case of allyl
bromide azidation should be carried out at 35-40°C for 6 h. The product was cooled to room temperature and
poured into cold water (200 mL). The organic layer was separated. The aqueous layer was extracted with benzene
(3x15 mL); the extracts were combined with the organic phase, dried (MgSO4), and filtered. Triethyl
phosphite (8.3 g, 0.05 mol) was added dropwise with stirring to a benzene solution of crude alkyl azide 1. The
temperature of the slightly exothermic reaction was kept at 25-30°C for 4 h and the product was left overnight
at room temperature. Benzene solution of N-alkyltriethoxyiminophosphorane 2 such prepared was added dropwise
with stirring and occasional external cooling (ice-water bath) to a solution of freshly distilled aldehyde
(0.05 mol) in benzene (5 mL) at 0-5°C. After the addition had been completed the mixture was stirred for 3 h
at 20-25°C. Benzene solution of crude imine 3 was then evaporated under reduced pressure end the residue was
diluted with methanol (100 mL). Sodium borohydride (1.9 g, 0.05 mol) was added portion-se with stirring to the
resultant solution and the temperature was kept below 30°C. The mixture was then left at room temperature far
24 h, evaporated under reduced pressure and diluted with water (50 mL). The solution was made strongly
alkaline With an excess of 50% aqueous sodium hydroxide and steam distilled. The distillate was acidified with
25% hydrochloric avid and evaporated to dryness. Crude amine hydrochloride 5 was dried in vacuo over P2O5 and
crystallized from the suitable solvent (see Table 1). When the amine could not be distilled with steam (entry 1)
the crude reduction product after evaporation of methanol was steam distilled to remove volatile contaminants.
The residue was made alkaline and extracted with ether (3x50 ml). The extracts were evaporated, the
residue was treated with 25% hydrochloric acid (10 mL), and evaporated to dryness to give crude 5. Analytically
pure samples were obtained by recrystallization from ethanol-ether unless otherwise stated (see Table 1).

Abstract
A new one-pot procedure for transforming primary alkyl bromides into the corresponding imines via the
Staudinger reaction has been developed. Acetonitrile was found to be an excellent solvent for azidation as
well as the reaction of organic azide with triphenylphosphine and a carbonyl compound.
It is well known that reaction of azides with tertiary phosphines gives iminophosphoranes1, useful
intermediates for the synthesis of a variety of nitrogen compounds2 including imines3. As part of our
program on the photochemistry of some allyl imines, we directed our efforts toward their efficient
preparation. Now we wish to report an effective one-pot synthesis of imines directly from alkyl bromides via
azide intermediates under the conditions of the Staudinger reaction in acetonitrile.
Scheme 1
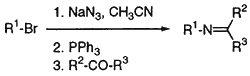
The literature reports a great number of procedures for the synthesis of alkyl azides by the nucleophilic
substitution of bromide with sodium azide4. To obtain a corresponding imine, isolated alkyl azides are
generally treated with triphenylphosphine and a carbonyl compound in a nonpolar solvent such as benzene3.
Those procedures are usually too complex and so we decided to find a short, simple, and efficient method.
Table 1
| Product | R1 |
R2 |
R3 |
Yielda |
| 1 | PhCH2 |
Ph | H | 95% (96%) |
| 2 | PhCH2 |
o-NO2-Ph |
H | 89% (98%) |
| 3 | PhCH2 |
n-Bu |
H | 94% (96%) |
| 4 | PhCH2 |
Ph | CH3 |
35%b |
| 5 | PhCH=CHCH2 |
Ph | H |
86% (70%)c |
| 6 | n-C9H19 |
Ph | H |
88% (94%)d |
Notes:
a. Yields of isolated compounds (purity shown in the
parenthesis was determined by NMR and/or by GC).
b. Refluxed 48 h after acetophenone addition, the
yield given is imine conversion by 1H-NMR.
c. 30% of unidentified byproducts were formed.
d. Azidation was carried out in wet acetonitrile.
Acetonitrile was found to be a very practical solvent in both steps: azidation and imine formation. Thus,
both steps were combined without necessity to isolate and purify any intermediate (Scheme 1). Reactive primary
bromides were refluxed with an excess of sodium azide in acetonitrile. Sodium bromide and the remaining sodium
azide were easily filtered off from the cooled reaction mixture because both salts are insoluble in cold
acetonitrile. Since aliphatic nonyl bromide did not afford nonyl azide under those conditions, a small amount
of water or a phase-transfer catalyst were used to enhance the reactivity. The presence of a phase-transfer
catalyst raised the conversion, however, the reaction time was too long (over 50 hours). Reflux of the
acetonitrile solution with triphenylphosphine and an aldehyde afforded the corresponding imine in a very high
yield and purity (Table 1). Reaction of cinnamyl bromide provided imine with only 70% purity - some unidentified
byproducts were formed. When ketone instead of aldehyde was used a longer reaction time in the second step was
applied bat the conversion was still too low.
The described one-pot procedure offers relatively high overall yields and thus may serve as an helpful
sequence in many procedures whom the synthesis of imines is anticipated.
Experimental
Synthesis of N-Benzyl-N-(phenylmethylidene)amine 1.
A stirred mixture of benzyl bromide (2.0 g, 11.7 mmol) and sodium azide (1.0 g, 15.2 mmol) in dry acetonitrile
(30 ml) was refluxed for 3 h and then cooled to 20°C Solid sodium bromide and remaining sodium azide were
removed by filtration and the benzyl azide solution was used in the next step without further purification.
Triphenylphosphine (2.8 g, 10-5 mmol) was added and the solution was refluxed for 1 h. Freshly distilled
benzaldehyde (1.1g, 10.5 mmol) was then added into the mixture and reflux continued for 2 h. Acetonitrile was
evaporated in vacuo and the residue was triturated twice with dry hexane (2x40 mL). The solid that precipitated
(triphenylphosphine oxide) was removed by filtration and concentration in vacuo gave imine 1. The analytically
pure imine was obtained by distillation (Kugelrohr).
Compounds 2, 3, and 5 were prepared according to this procedure from the corresponding bromides and
aldehydes. Synthesis of imine 4 was same as above besides that the acetonitrile solution was refluxed for 48 h
after the addition of acetophenone.
Synthesis of N-Nonyl-N-(phenylmethylidene)amine 6.
A stirred mixture of nonyl bromide (2.0 g, 9.7 mmol) and sodium azide (0.8 g, 12.6 mmol) in a mixture of
acetonitrile (27 ml) and water (3 ml) was refluxed for 12 h. and then cooled to 20°C. The water layer was
removed and the acetonitrile solution was dried with sodium sulfate. The filtered solution was used in the
next step without further purification and was treated with triphenylphosphine and benzaldehyde in the same
way as that described in the first procedure.
- N-Benzyl-N-(phenylmethylidene)amine 1: bp 195-200°C/10 Torr.
- N-Benzyl-N-[(2-nitrophenyl)methylidene]amine 2: bp 220-225°C/10 Torr.
- N-Benzyl-N-(butylidene)amine 3: bp 110-115°C/10 Torr.
- Phenyl-N-(1-phenylethylidene)methanamine 4: The compound was not isolated.
- N-(Phenylmethylidene)-N-[(E)-3-phenylprop-2-enyl]amine 5: bp 120-135°C/0.05 Torr.
- N-Nonyl-N-(phenylmethylidene)amine 6: bp 175-185°C/10 Torr.
Abstract
This paper presents a high yielding one-pot solution phase and polymer supported synthesis of a range of primary and secondary amines starting from azides and aldehydes. The synthesis utilises a tandem process which begins with an aza-Wittig reaction between the aldehyde and an iminophosphorane, followed by reduction, or organometallic 1,2-addition reaction, of the resultant imine. The requisite iminophosphoranes were accessed using the highly efficient Staudinger reaction between the azide starting material and a phosphine. The process was applicable to the solid phase by the use of polymer supported iminophosphoranes and polymer supported cyanoborohydride.
The conversion of aldehydes and ketones into imines and the subsequent synthesis of α-unsubstituted and a-branched secondary amines by the reduction of the imine,1,2 by the 1,2-addition of organometallic reagents to the imine,3,4 or by the addition of other nucleophiles to the imine5 is of current and continuing importance. Several recent reports have been concerned with the use of solid phase methods for achieving such transformations.6-8 The use of azides as the nitrogen source for the synthesis of primary amines can be achieved by the reduction of the azide or by the conversion of the azide into an iminophosphorane [the Staudinger reaction] followed by hydrolysis,9 a process which has been transferred to the solid phase.10 These methodologies do not involve imine intermediates and cannot be used to access secondary amines. The solution phase conversion of azides into Boc-protected amines has been reported,11 and other methods for the conversion of azides into secondary amines which do not involve iminophosphorane or imine intermediates are also known.12 We are aware of only a few isolated specific examples for the solution phase conversion of azides into secondary amines,13a particularly polyamines,13b via intermediate iminophosphoranes and imines. However, these reports do not disclose widespread applicability and no detailed or general studies have appeared to date. No solid phase applications for the conversion of azides into α-unsubstituted or α-branched secondary and primary amines via intermediary imines and iminophosphoranes have been reported.14
Scheme 1
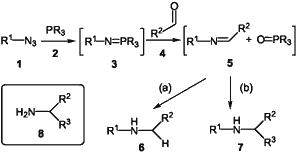
- NaBH3CN or NaBH4 or BH3-THF; MeOH, NH4Cl(aq)
- R3MgX or RLi; NH4Cl(aq)
In this preliminary report, we present a simple, general, one-pot solution phase and polymer supported protocol for the combination of azide and aldehyde starting materials which allows access to α-unsubstituted and α-branched secondary and primary amines. The chemistry proceeds via the in situ reactions of intermediate iminophosphoranes and imines, where the iminophosphoranes may be polymer supported. The solid phase process offers significant advantages over the corresponding solution phase method in terms of time, ease of purification, avoidance of chromatography and cost. A clear advantage of the solid support methodology is its potential for the generation of combinatorial libraries of amines using readily available starting materials which offer several points of diversity.
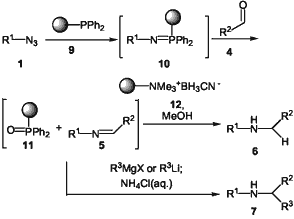
Scheme 2
We began our study by establishing the validity of the chemistry in the solution phase, as shown in Scheme 1. Thus, the Staudinger reaction15 between the phosphine 2 and the azide 1 gave the iminophosphoranes 3, which were then made to undergo in situ aza-Wittig reaction16 with aldehydes 4 to give the imines 5. The in situ reduction of the imines 5 with sodium borohydride, sodium cyanoborohydride or borane-THF gave, after flash silica chromatography, the α-unsubstituted amines 6, generally in high yields as shown in the Table. The in situ reaction of the imines 5 with an organometallic (R3Li or R3MgX) similarly allowed access to the α-branched amines 7, again in high yields. The range of suitable organometallic reagents included allyl magnesium bromide, thereby allowing access to synthetically useful homoallylic amines.4a,7a,17 The use of benzyl azide allowed a practical synthesis of N-benzyl protected amines whilst the use of trimethylsilyl azide (1, R = TMS) allowed access to primary amines 8, a process which proceeds via the in situ N-desilylation17 of the intermediate N-silylated amines4a,17 upon workup.
Table
Preparation of Amines From Azides & Aldehydes
|
R1 |
R2 |
R3
[see text] |
|
|
1 | TMS- | Ph- | Allyl- |
87% |
79% |
2 | TMS- | p-MeO-Ph- | Allyl- |
81% |
64% |
3 | TMS- | p-MeO-Ph- | n-Bu- |
74% |
69% |
4 | TMS- | p-MeO-Ph- | Ph- |
71% |
68% |
5 | TMS- | Ph- | Ph- |
55% |
59% |
6 | PhCH2- | Ph- | H- |
97% |
>99% |
7 | PhCH2- | PhCH=CH- | H- |
93% |
>99% |
8 | PhCH2- | t-Bu- | H- |
20% |
|
9 | PhCH2- | Ph- | Allyl- |
84% |
91% |
10 | PhCH2- | Ph- | n-Bu |
76% |
83% |
11 | Ph- | Ph | H- |
>99% |
|
12 | Ph- | PhCH=CH- | H- |
87% |
|
13 | Ph- | Ph | Allyl- |
91% |
95% |
14 | Ph- | Ph | n-Bu |
>99% |
>99% |
15 | Ph- | p-F-Ph- | H- |
75% |
|
16 | Ph- | n-Pr | H- |
66% |
|
17 | Ph- | Me | H- |
58% |
|
18 | Ph- | PhCH=CH- | Bu- |
92% |
96% |
19 | Ph- | Me(CH=CH)2- | H- |
70% |
58% |
20 | p-MeO-Ph- | Ph | H- |
>99% |
91% |
- All compounds gave spectral data consistent with the
assigned structure and agreeing with lit. values2,4,7,17
- Solution-phase. Yield of pure, isolated compound.
- Polymer Supported. Yield of pure, isolated compound.
- Reduction performed with BH3CN- on polymer support21
The Table shows the azides, aldehydes and organometallics that were used in our study, together with the isolated yields of the amines. In the solution phase, we have also screened the use of a range of trivalent phosphorus species 2. We found that the use of tri(n-butyl)phosphine typically gave yields of amines 5-10% higher than triphenylphosphine. The use of trimethyl phosphite, triethyl phosphite and hexamethylphosphorous triamide allowed the process to be carried out at lower temperatures than with trialkyl- or triaryl-phosphines without significant differences in yield. However, we believe that ease of handling, odour containment and other safety issues make triphenylphosphine the phosphine of choice for this process, and the Table refers to results obtained with this phosphine.18
Gratifyingly, all stages of this chemistry were compatible with transfer to a polymer supported protocol by the use of polymer supported triphenylphosphine19 at the phosphine stage to give the polymer supported iminophosphorane 10 as shown in Scheme 2.20 Advantageously, we found that polymer supported cyanoborohydride brought about the reduction of the intermediate imines 5 in good yields, as shown in the Table.20,21 Chromatographic separation of the phosphine oxide was not required: the polymer supported phosphine oxide 11 was removed by filtration. Polymer supported phosphine 9 can be regenerated by the reduction of polymer supported phosphine oxide 11 with trichlorosilane.19a The use of Amberlite® azide exchange resins for the synthesis of azides and the availability of polymer supported methods for the synthesis of aldehydes from alcohols8a offer further possibilities which may be significant for future applications in combinatorial chemistry.
In conclusion, we have developed a simple polymer supported method for the combination of azides with aldehydes to give α-unsubstituted and α-branched primary and secondary amines. Further studies on the development of methodologies for the polymer supported asymmetric synthesis of α-branched amines and the polymer supported synthesis of nitrogen containing heterocycles based upon Staudinger/aza-Wittig chemistry are underway.