Abstract
An efficient O-alkylation of alcoholic tosylates with amines in a K2CO3/CO2 system in the presence of tetrabutyl ammonium iodide (TBAI) provides exclusive formation of carbamates.
Organic carbamates represent an important class of compounds having various properties. They have largely found use as agrochemicals1a,b (pesticides, fungicides, herbicides), pharmaceuticals2a,2b as intermediates in organic synthesis3, protecting groups in peptide synthesis4 and as linkers in combinatorial chemistry5a,b. Hence considerable interest has generated in the recent past for development of an effecient and safe methodology for carbamate synthesis. Classical syntheses of carbamates involve direct alcoholysis of phosgene or its derivatives6, aminolysis of chloroformates7 and alcoholysis of isocyanates8, thus utilizing phosgene, a harmful chemical, directly or indirectly.
Recently carbondioxide, a cheap and safe reagent, has been tapped as an alternative to phosgene for the introduction of a CO group. Preparation of carbamates has been reported using CO2 electrochemically9a,9b, super critically10 and in combination of metal and non-metal species11a-d. CO2 alone has low reactivity with nucleophiles. With amines it gives unstable carbamic acid (1). However, with 2 molar equivalence of amine, monoalkylammonium alkyl carbamate ion (2) is formed.
Scheme 1
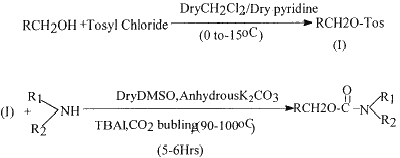
The alkylammonium carbamate thus formed reacts with alkyl halide to give rise to N- or O-alkylation (carbamate) products. Formation of O-alkylation product has been reported in the reaction of primary or secondary amines, CO2 and alkyl halides in the presence of strong proton acceptors e.g., DBU12, transition metal complexes and crown ether13.
It was observed14-16, that presence of a phase transfer catalyst in the alkylation of secondary amines with various alkyl halides led to the formation of carbamates.
Entry No. |
R |
R1 |
R2 |
Time |
Yields |
1 | n-C3H7 | C8H17 | H |
5–6 h |
90.50% |
2 | (H3C)2CHCH2 | C8H17 | H |
5–6 h |
73.16% |
3 | H3C(CH2)3 | C4H9 | H |
5–6 h |
75.10% |
4 | H3C(CH2)4 | Cyclohexyl | H |
5–6 h |
88.25% |
5 | H3C(CH2)4 | C3H7 | H |
5–6 h |
72.26% |
6 | H3C(CH2)6 | 3-MePhCH2 | H |
5–6 h |
90.03% |
7 | H3C(CH2)8 | C6H13 | H |
5–6 h |
82.60% |
8 | PhCH2 | C4H9 | H |
5–6 h |
88.65% |
9 | PhCH2CH2 | C6H13 | H |
5–6 h |
80.06% |
10 | PhCH2 | C3H7 | C3H7 |
5–6 h |
71.13% |
Based on the concept of formation of the ionic species 2, we investigated the carbamate synthesis from alcoholic tosylates. The carbonium ion generated from the tosyl esters would undergo nucleophilic attack by 2 leading to the formation of carbamate esters (Scheme 1). Thus, tosyl ester of an alcohol which is easily synthesized17 by reacting alcohol with p-toluene sulphonyl chloride was used as a substrate for the preparation of carbamates. Different alcoholic tosylates were reacted with the aliphatic/aromatic amines in dry DMSO in the presence of CO2/K2CO3 and a phase transfer catalyst, tetrabutyl ammonium iodide, to furnish desired carbamate esters in high yields. The order of yield of carbamates formed from amines was found to be as follows: RNH2>R2NH>Ar-NH2. In the case of aromatic amines, use of benzyl triethyl ammonium hydroxide (Triton-B) as phase transfer catalyst and dry HMPA as a solvent gave better results. The yields of various carbamates prepared by using different amines and alcoholic tosylates is given in the Table 1.
Experimental
Typical Method Of Preparation
n-Butyl n-octylcarbamate (1): A mixture of anhydrous K2CO3 (5g), dry DMSO (30 mL), n-octylamine (0.72 mL, 4 mmole), was taken in a 50 mL two neck flask. The temperature of the reaction was maintained at 90–100°C and purified CO2 gas was bubbled through it. Reaction was continued for 1h, tetrabutylammonium iodide (0.20g, 5 mmole) was added to it. The reaction was further continued for ½h, n-butyltosyl ester (0.5g, 2.19 mmole) was added to it and reaction continued for another 3–4h. The completion of the reaction was checked by TLC. Reaction mixture was poured in to distilled water (50 mL) and extracted with ethylacetate thrice. The organic layer was dried over anhydrous Na2SO4 and concentrated to get pure oily compound (Yield 0.43 g, 90.52%, oil).
IR: 1787 cm-1 (O-CO-NH). 1H-NMR (CDCl3): δ 0.77–0.87 (t, 6H, CH3), 0.89–0.99 (m, 4H, CH2), 1.02–1.21 (m, 4H, CH2), 1.25–1.34 (m, 4H, CH2), 1.35–1.39 (m, 2H, CH2), 1.52–1.54 (m, 2H, CH2), 3.87–3.92 (t, 2H, CH2), 4.02–4.06 (t, 2H, CH2O), 4.67–4.71 (bs, H, NH).
Isoamyl n-Octylcarbamate (2): IR (Neat) = 1702 cm-1 (-O-CO-NH). 1H NMR (CDCl3) = 0.67–0.75 (d, 6H, CH3), 0.85–0.93 (t, 3H, CH3), 0.95–1.34 (m, 12H, CH2), 1.35–1.37 (m, H, CH), 1.7–1.9 (m, 2H, CH2), 4.02–4.04 (t, 2H, CH2), 4.73–4.75 (bs, H, NH). (Yield 0.36 g, oil).
n-Pentyl n-butylcarbamate (3): IR (Neat) = 1789 cm-1 (O-CO-NH). 1H NMR (CDCl3) = 0.87–0.95 (t, 6H, CH3), 1.02–1.66 (m, 12H, CH2), 4.05–4.21 (t, 2H, OCH2), 4.65–4.73 (bs, H, NH), (Yield, 0.29 g, oil).
n-Hexyl cyclohexylcarbamate (4): IR (KBr) = 1698 cm-1 (O-CO-NH), 1H NMR (CDCl3) = 0.85–0.91 (t, 3H, CH3), 0.95–1.71 (m, 6H, CH2), 4.02–4.06 (t, 2H, CH2-O), 4.75–4.78 (bs, H, NH), 1.99–2.10 (m, 10H, cyclohexyl-CH2), 1.91–2.1 (m, CH of cyclohexyl ring) (Yield 0.43 g, m.p. 139–140°C).
n-Hexyl n-propylcarbamate (5): IR (Neat) = 1693 cm-1 (O-CO-NH), 1H NMR (CDCl3) = 0.85–0.93 (m, 3H, CH3), 1.22–1.66 (m, 12H, CH2), 2.3–2.7 (s, 2H, CH2), 3.7–3.7 (s, 3H, OCH3), 4.73–4.77 (bs, H, NH), 7.12–7.77 (m, 4H, Ar-H) (Yield, 0.26 g, oil).
n-Octyl m-methoxybenzylcarbamate (6): IR (KBr) = 1705 cm-1 (O-CO-NH) 1H NMR (CDCl3) = 0.87–0.93 (t, 6H, CH3), 1.02–1.77 (m, 10H, CH2), 3.22–3.55 (m, 2H, CH2-NH), 4.05–4.12 (t, 2H, CH2) (Yield 0.45 g, m.p. 137°C).
n-Decyl n-hexylcarbamate (7): IR (Neat): 1693 cm-1. 1H NMR (CDCl3) = 0.89–0.93 (m, 12H, CH2), 1.55–1.67 (m, 8H, CH2), 2.96–2.99 (t, 2H, CH2), 4.10–4.32 (t, 2H, CH2-O), (Yield 0.38 g, oil).
2-Phenylethyl n-butylcarbamate (8): (18) IR(neat): 1692 cm-1 (O-CO-NH). 1H NMR (CDCl3) = 0.87–0.94 (3H, m, CH3), 1.25–1.35 (2H, m, CH2), 1.38–1.44 (2H, m, CH2), 2.13–2.20 (2H, m, CH2NH), 2.3–2.7 (2H, m, CH2Ar), 3.5–3.8 (2H, t, CH2O), 7.12–7.77 (5H, m, Ar-H) (Yield 0.37 g, oil).
3-Phenylpropyl n-hexylcarbamate (9): IR (Neat) = 1697 cm-1 (O-CO-NH). 1H NMR (CDCl3) = 0.92–0.97 (t, 3H, CH3), 1.29–1.32 (m, 10H, CH2), 2.93–2.85 (t, 2H, CH2-N), 2.87–2.95 (t, 2H, CH2-Ar), 4.22–4.32 (t, 2H, CH2), 7.12–7.77 (m, 5H, Ar-H) (Yield 0.38 g, oil).
2-Phenylethyl di-isopropylcarbamate (10): IR (Neat) = 1695 cm-1 (-O-CO-NH). 1H NMR (CDCl3) = 0.89–0.94 (t, 6H, CH3), 1.23–1.44 (m, 8H, CH2), 2.83–2.86 (t, 4H, CH2), 2.87–2.95 (t, 2H, CH2-Ar), 4.21–4.32 (t, 2H, CH2-O), 7.13–7.77 (m, 5H, Ar-H) (Yield, 0.38 g, oil).