Abstract
In the past, it has been argued in court, from a theoretical basis, that the techniques available to the forensic chemist would differentiate the "cocaines". This work has moved that argument from the realm of the theoretical into that of experimental fact. The techniques of infrared spectroscopy (IR), nuclear magnetic resonance (NMR), and mass spectrometry (MS) will unequivocally identify the racemic cocaine diastereoisomer. In addition, this work shows that the enantiomeric form of cocaine can be assigned by crystal tests, IR, and melting point techniques. The pure enantiomers of allococaine and pseudoallococaine were not isolated. This does not create a problem because the techniques of NMR and MS, as performed in this study, will not differentiate enantiomers. Therefore, the logical sequence of first identifying the diastereoisomer (via IR, NMR, or MS) and then determining the chirality by crystal tests, IR, melting points, or optical rotation measurements is valid.
Fig. 1.
Diastereoisomers of 2-carbomethoxy-3-benzoyloxytropane
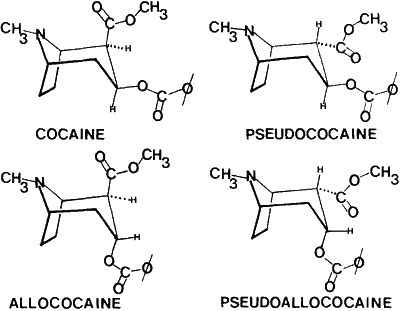
Questions have arisen recently in court concerning the specificity of instrumental and chromatographic techniques for the differentiation of the diastereoisomers of cocaine (Fig. 1). This paper presents analytical techniques that can be used for the differentiation of these diastereoisomers and the optical isomers of cocaine. The techniques presented are melting points (MP), thin-layer chromatography (TLC), high pressure liquid chromatography (HPLC), gas-liquid chromatography (GLC), microcrystalline tests, nuclear magnetic resonance spectrometry (NMR), infrared spectroscopy (IR), and electron impact mass spectrometry (MS).
Procedure
The diastereoisomeric "cocaines" were synthesized as described in the literature1-7. Melting points were obtained with the last 10°C traversed at 2°C/min and were corrected by comparison to a standard curve. A Thomas Hoover capillary melting point apparatus and borosilicate glass capillaries were used. An iodine tank was used for visualization and Kontes equipment (Quanta Gram®) for the TLC work. The HPLC work was performed on a Waters Associates instrument with an M6000A delivery system with an ultraviolet (UV) detector. The GLC work was performed on Packard 7300, Hewlett-Packard 5840, and Finnigan 9500 gas chromatographs, all with heated injection ports and fitted with glass columns. Microcrystalline work was accomplished with a Zeiss Standard WL Research microscope adapted for microphotography. The microcrystalline work was performed as suggested by Fulton8. The NMR spectra were obtained on the free bases in deuterated chloroform, with tetramethylsilane as reference, with a Varian EM 390 (90 MHz). A Finnigan Model 3300 GC/MS interfaced with the Finnigan 6110 data system was used for producing the mass spectra. Infrared spectra were obtained from potassium bromide disks with a Perkin-Elmer 283 grating spectrophotometer.
Results
Table 1.
Melting point data °C.
| Compound |
Findlay4
|
Sinnema
et al.5 |
Willstätter
et al.7 |
Merck9
|
This
Work |
| (-)-Cocaine |
-
|
-
|
-
|
98
|
96-97
|
| (+)-Cocaine |
-
|
-
|
98
|
-
|
-
|
| (±)-Cocaine |
79-80
|
-
|
79-80
|
-
|
74-75
|
| (-)-Cocaine HCl |
-
|
-
|
192
|
195
|
194-196
|
| (±)-Cocaine HCl |
187
|
-
|
187
|
-
|
186-188
|
| (+)-Pseudococaine |
-
|
-
|
43-45
|
47
|
-
|
| (±)-Pseudococaine |
81.5
|
-
|
81.5
|
-
|
oil
|
| (+)-Pseudococaine HCl |
-
|
-
|
205
|
210
|
208-210
|
| (±)-Pseudococaine HCl |
205.5
|
-
|
205-206
|
-
|
202-204
|
| (±)-Allococaine |
93-95
|
95-97
|
-
|
-
|
94-96
|
| (±)-Allococaine HCl |
oil
|
oil
|
-
|
-
|
oil
|
| (±)-Pseudoallococaine |
82-84
|
83-84
|
-
|
-
|
oil
|
| (±)-Pseudoallococaine HCl |
201.5
|
209-210
|
-
|
-
|
203-205
|
Melting Points
Table 1 presents the melting point data for the diastereoisomeric cocaines obtained by Findlay4, Sinnema et al.5, Willstätter et al.7, and Merck & Co.9 as well as our results. In this work, values for the bases of pseudococaine, pseudoallococaine, and the hydrochloride salt of allococaine are not included. These compounds were obtained as clear colorless oils.
Thin-Layer Chromatography
Table 2 presents Rf values for the four diastereoisomers obtained with silica gel as the stationary phase and acetonitrile as the mobile phase. The developing distance was approximately 18 cm.
Table 2.
Rf values of the four
diastereoisomers on
SiO2 eluting w. MeCN
| Compound | Rf |
| Cocaine | 0.19 |
| Pseudococaine | 0.39 |
| Allococaine | 0.42 |
| Pseudoallococaine | 0.25 |
Table 3.
Results obtained by HPLC
on a Partisil 10 PAC column
eluted with acetonitrile.
| Compound |
Retention
Volume |
| Cocaine |
33 mL
|
| Pseudococaine |
78 mL
|
| Allococaine |
43 mL
|
| Pseudoallococaine |
50 mL
|
HPLC
A Partisil 10 PAC® column (Whatman) eluted with acetonitrile produced the results shown in Table 3.
GLC
Table 4 lists retention times Rf and retention times relative to cocaine (RR) for the free bases of the diastereoisomeric cocaines on a packed 3% OV-1 column in an all-glass system.
Table 4.
Results obtained by GLC.
| Compound |
Retention Time
|
|
|
Absolute
|
Relative
|
|
| Cocaine |
3.27
|
1.0
|
| Pseudococaine |
3.61
|
1.1
|
| Allococaine |
4.2
|
1.3
|
| Pseudoallococaine |
2.98
|
0.9
|
Carrier gas: He, 30 mL/min; 200°C;
Column: 0.2×100cm 3% OV-1
Silicone liquid phases of intermediate polarity (OV-17, OV-25) did not adequately resolve the cocaine diastereoisomers. The low-polarity silicone phases (OV-1, OV-101) performed considerably better, although injection of a mixture of cocaine hydrochloride and pseudococaine hydrochloride resulted in a single peak with a retention time intermediate to those given for cocaine and pseudococaine.
Microcrystalline Tests
Microcrystalline tests were performed on the racemic mixtures of all four cocaine diastereoisomers and on (-)-cocaine and (+)-pseudococaine. Salts of platinum (5% platinic chloride in 3N hydrochloric acid) and gold (3-5% acid gold chloride in 25% acetic acid) were used as precipitating reagents. Cocaine is the only one of the four diastereoisomers to give a crystalline precipitate with the gold reagent. These reagents also provide unequivocal assignment of the enantiomeric form of cocaine. The method involves the simple addition of a known enantiomer to an unknown as described by Fulton10 and Clarke11:
- (-)-cocaine + (-)-cocaine + acid gold chloride = (-)-crystals (Fig. 2)
- (+)-cocaine + (-)-cocaine + acid gold chloride = (±)-crystals (Fig. 3)
{kind=link}
{kind=link}
Fig. 2 and Fig. 3 are photomicrographs of a (-)-crystal and (±)-crystals obtained with acid gold chloride in acetic acid. Note that the (±)-crystals are at least partially amorphous.
Platinic chloride (H2PtCl4) in 3N hydrochloric acid reacted with (-)-cocaine and gave crystals very similar to those described by Fulton12.
Racemic cocaine and platinic chloride in 3N hydrochloric acid, unlike gold chloride, does give a completely crystalline precipitate. These crystals are quite small (<50 µm) and appear to be plates arranged around a central point. Under crossed polarizers, the single color of yellow is present with moderate intensity.
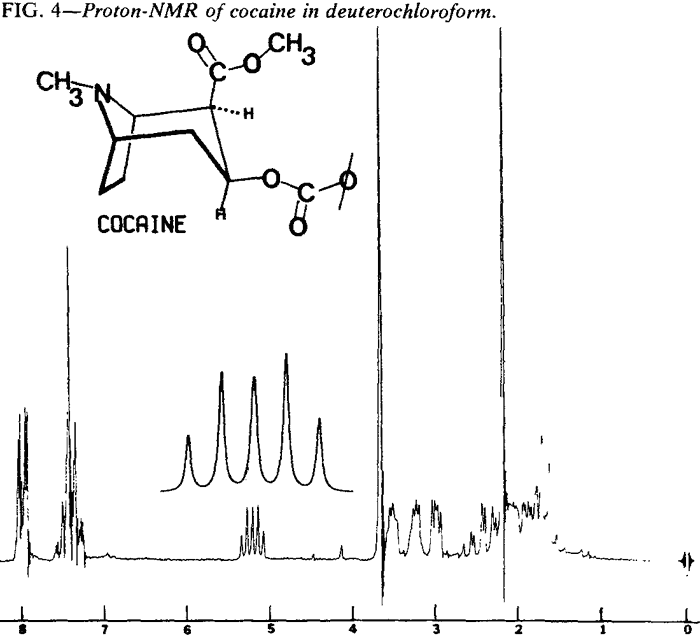
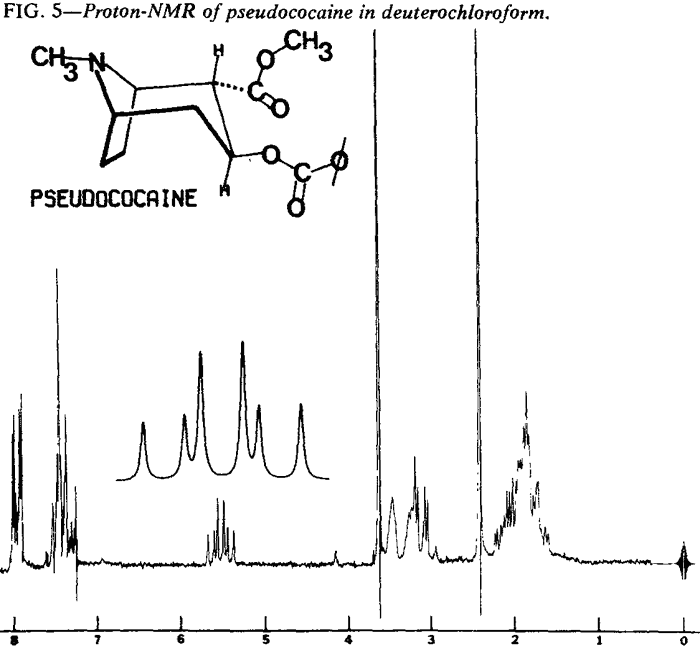
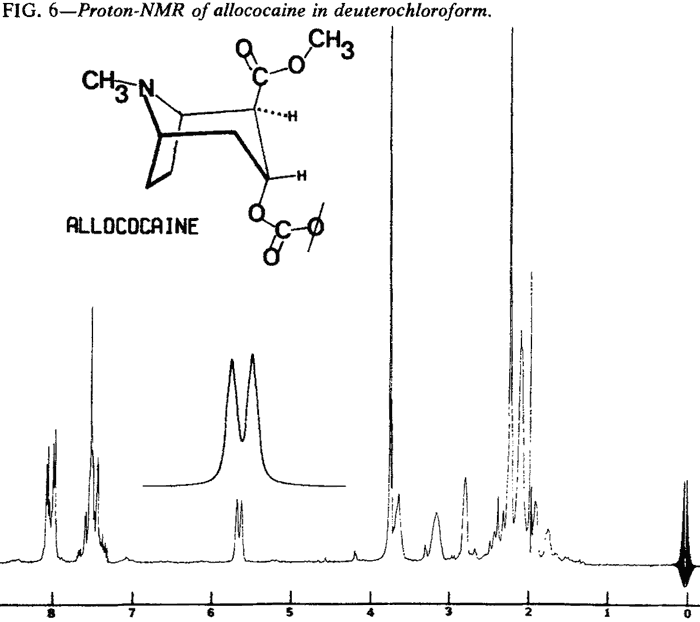
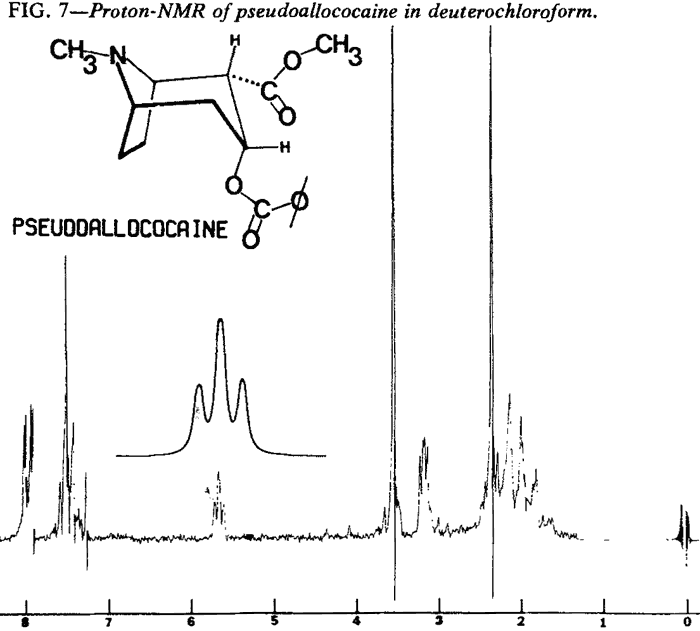
Proton Nuclear Magnetic Resonance
| Fig. 4. | Cocaine |
| Fig. 5. | Pseudococaine |
| Fig. 6. | Allococaine |
| Fig. 7. | Pseudoallococaine |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Proton NMR spectra of the diastereoisomeric cocaines are significantly different. Major differences are seen in the chemical shifts associated with the respective C-3 protons. In addition, the coupling patterns that arise from the vicinal coupling of C-2 and C-4 protons with the C-3 proton are first order and relate nicely to the Karplus equation5.
That the diastereoisomeric cocaines, can easily be distinguished by observation of the chemical shifts and coupling patterns associated with the C-3 proton does not imply that other differences do not exist. In fact, virtually every proton in these molecules exhibits a different chemical shift or coupling pattern (Figs. 4-7).
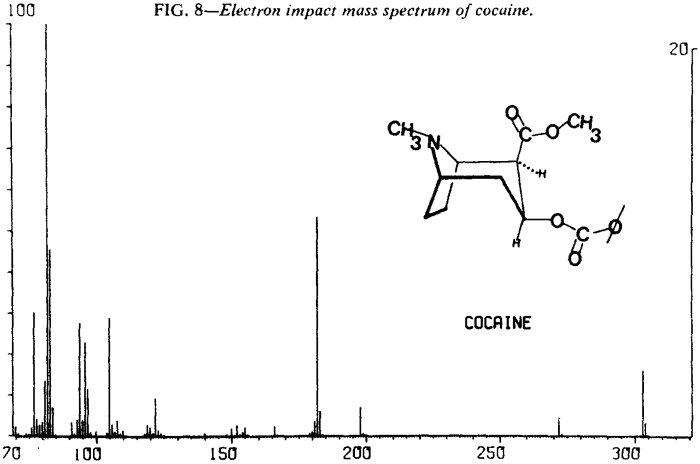
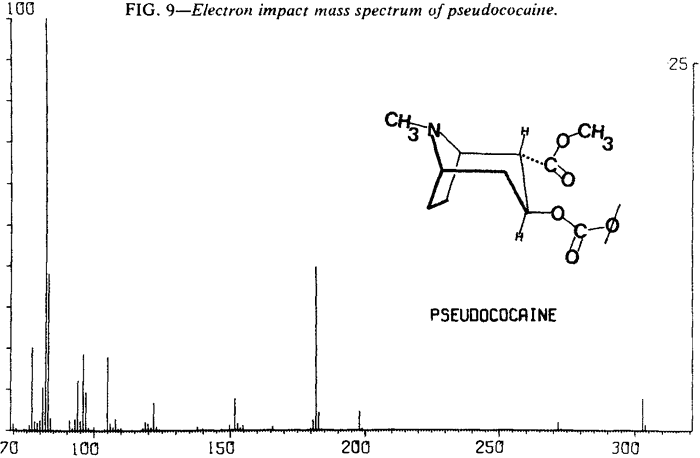
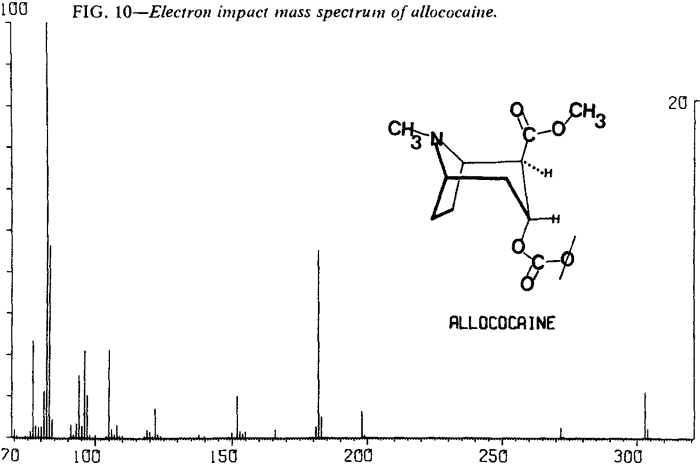
Mass Spectrometry
| Fig. 8. | Cocaine |
| Fig. 9. | Pseudococaine |
| Fig. 10. | Allococaine |
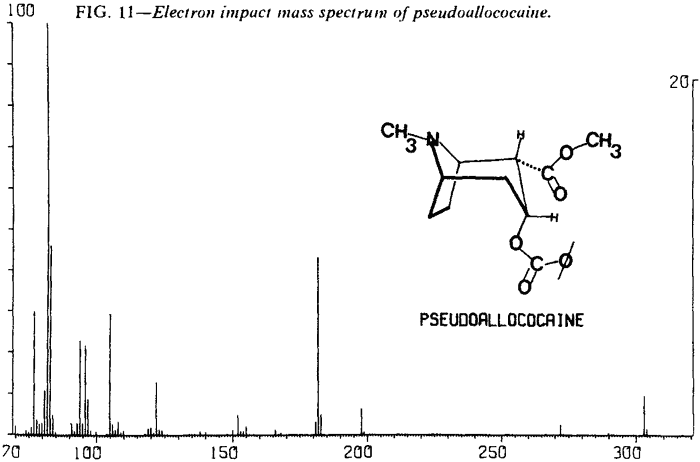
| Fig. 11. | Pseudoallococaine |
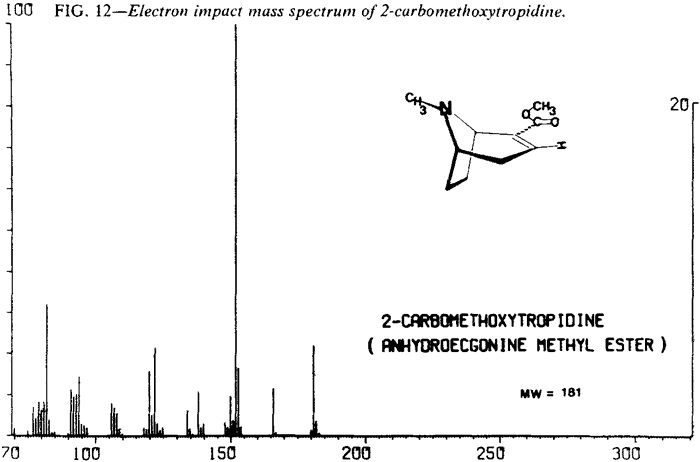
| Fig. 12. | 2-Carbomethoxytropidine |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
As expected, low resolution electron impact mass spectra of the diastereoisomeric cocaines are similar. Fortunately, cocaine and pseudoallococaine can be differentiated from each other and the other two diastereoisomers. The ions that accomplish this differentiation most easily are at m/e 94, 96, 150, and 152. The stereochemistry of the C-2 carbomethoxy and the C-3 benzoyloxy results in varying abundances of the ions at m/e 94 and m/e 15213. In addition, examination of the mass spectra shows that the relative abundances of ions at m/e 96 and m/e 150 remain nearly constant (Figs. 8-11). This allows the ratios of m/e 94:96 and m/e 152:150 to be used for the differentiation of these compounds and thereby removes the need for a normalized spectrum.
Table 5 lists abundance ratios for these ions at 70 eV. These ratios were confirmed with the following experimental conditions and instruments:
Table 5.
Abundance ratios
| Compound |
m/e
94:96 |
m/e
152:150 |
| Cocaine |
>1
|
1-2
|
| Pseudococaine |
<1
|
5-7
|
| Allococaine |
<1
|
7-10
|
| Pseudoallococaine |
>1
|
3-5
|
- Finnigan 3000, 70 eV, source 60°C
- Finnigan 3300, 40 to 80 eV, source 60°C
- Finnigan 4000, 40 to 80 eV, source 150 to 240°C
- AEI MS-902, 70 eV, source 150 to 225°C
- AEI MS-30, 70 eV, source 200°C
These abundance ratios may not be sufficient for differentiation of allococaine from pseudococaine because of the possible overlap of m/e 152:150 ratios. In that case, relative abundances of other ions can be compared. For pseudococaine, the ions at m/e 105, 122, 182, 198, and 272 are at a lower relative abundance and the molecular ion (m/e 303) is at a higher relative abundance than the corresponding ions of allococaine.
The mass spectral data for the cocaines were obtained by direct insertion techniques rather than via a GLC interface. This was necessitated by a tendency of the less stable diastereoisomers, in particular pseudoallococaine, to thermally eliminate the elements of benzoic acid. The product of this thermal elimination is 2-carbomethoxytropidine (anhydroecgonine methyl ester). The electron impact fragmentation of this compound results in a spectrum with an m/e 152 base peak (Fig. 12). Since the differentiation of the diastereoisomeric cocaines relies heavily on the relative abundance of the ion at m/e 152, thermal elimination in the GLC/MS interface could interfere with that assessment.
Infrared Spectroscopy
| Fig. 13. | Cocaine HCl |
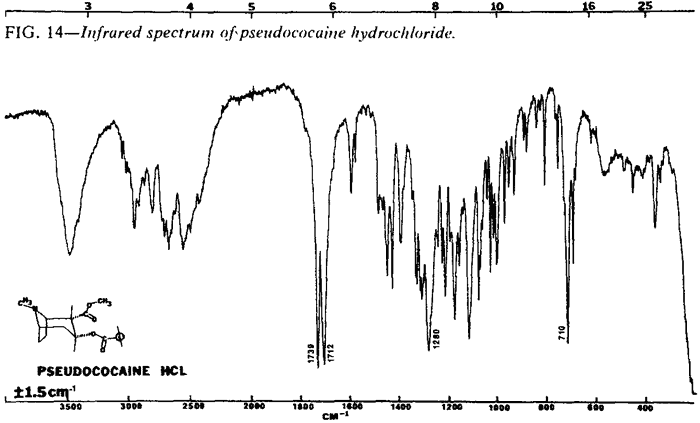
| Fig. 14. | Pseudococaine HCl |
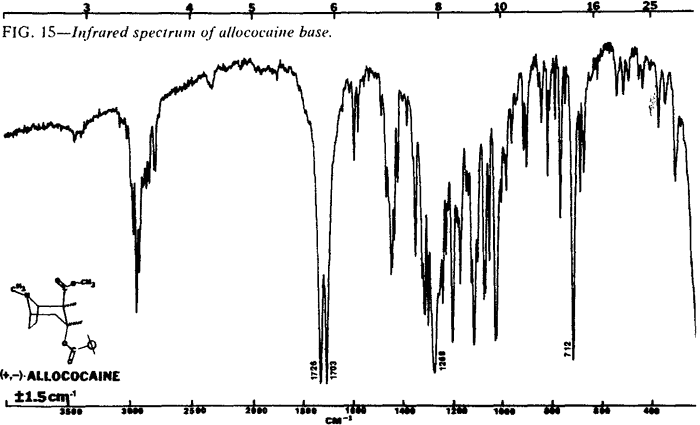
| Fig. 15. | Allococaine base |
| Fig. 16. | Pseudoallococaine HCl |
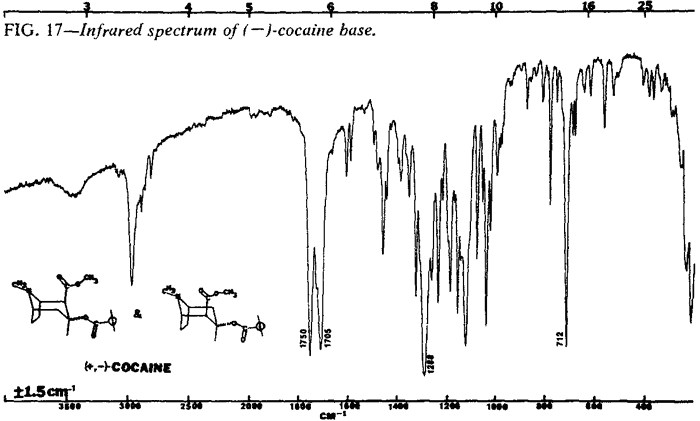
| Fig. 17. | (-)-Cocaine base |
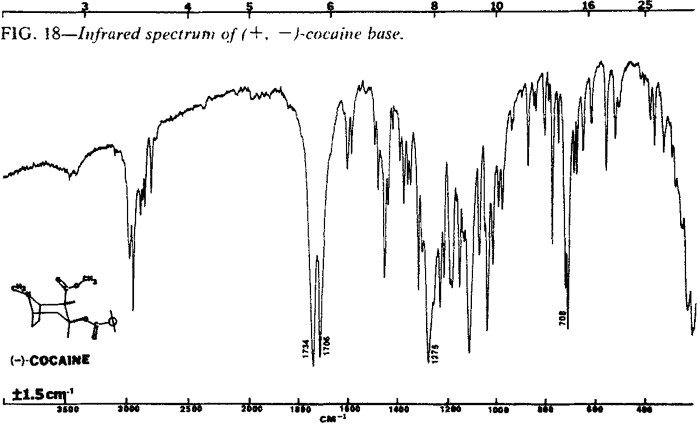
| Fig. 18. | (±)-Cocaine base |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The IR spectra of the cocaine diastereoisomers are shown in Figs. 13-18. Fig. 13 represents the IR spectrum of both (-) and (±)-cocaine hydrochloride because the spectra are identical. This occurs because cocaine hydrochloride, as both the pure enantiomer (-) and the racemic mixture (±), exists as a conglomerate or eutectic mixture. The IR spectra of the pure enantiomer (+)-pseudococaine hydrochloride and the racemic mixture (±) of pseudococaine hydrochloride are also identical for the same reason, and again are represented by only one spectrum (Fig. 14). However, the free bases of (±) and (-)-cocaine exhibit vastly dissimilar IR spectra (Fig. 17 and Fig. 18). In this case, the racemic mixture (±) forms a true racemate association. This was readily verified by melting point techniques14.
Discussion
Melting Points
Since the melting ranges of some of the diastereoisomers are very close, or even overlap, the melting point is unsatisfactory for the identification of the diastereoisomer present. However, since (-)-cocaine base and (±)-cocaine base have widely separated melting ranges, enantiomeric identification can be made with mixed melting point techniques14.
Chromatography
Of the chromatographic techniques tested, resolution of the cocaine diastereoisomers is best accomplished by HPLC, followed by TLC, then GLC. The main weakness of GLC is the tendency of these diastereoisomers to undergo thermal elimination, thus causing serous loss of resolution.
Microcrystalline Tests
The advantages of the microcrystalline technique are speed, selectivity, and sensitivity. In a very short time, the experienced chemist can identify both the cocaine diastereoisomer and the enantiomeric form on microgram quantities of material. The principal disadvantages of this technique are that the presence of other compounds in the sample can distort the microcrystalline precipitate and that the technique requires a certain degree of expertise on the part of the chemist.