57% Hydriodic acid1
A 1.5 litre three-necked flask is charged with a mixture of 480g of iodine and 600 ml of water. The central aperture is fitted with a stopper carrying an efficient mechanical stirrer leading almost to the bottom of the flask, and the smaller apertures respectively with a lead-in tube for hydrogen sulphide extending to well below the surface of the liquid and with an exit tube attached to an inverted funnel just dipping into 5% NaOH. The mixture is vigorously stirred, and a stream of hydrogen sulphide* is passed in as fast as it is absorbed. After several hours the liquid assumes a yellow colour (sometimes even colorless) and most of the sulphur sticks together in the form of a hard lump. The sulfur is removed by filtration through a funnel plugged with glass wool (or through a sintered glass funnel). The hydriodic acid is then distilled, and the fraction boiling between 125.5-126.5°C is collected as 57% hydrogen iodide. The yield is 90% of the theorethical.
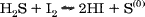
* Generated by dripping HCl on lumps of FeS (made by fusing iron powder with sulfur).
Anhydrous HI Gas
Hydrogen iodide may be conveniently prepared by allowing a solution of two parts of iodine in one part of hydriodic acid (d. 1.7), to drop onto an excess of red phosphorous. The reaction takes place in the cold. When the evolution of gas slackens considerably, the mixture should be gently warmed.
11 parts (by weight) of iodine is placed in a small flask, and 1 part of yellow phosphorous, cut into small pieces and dried, is gradually added. Expect a flash of light and the contents to turn liquid upon the addition. When all the phosphorous has been added, phosphorous triiodide is to be separated upon cooling. The product is treated with 1.5 parts water, heated gently to produce hydrogen iodide, which is passed over some red phosphorous, that has been moistened with a little water and placed in a U tube. Heating is continued until the liquid just becomes colorless, because if heating is continued further, phosphine and phosphonium iodide are formed, which can cause a powerful explosion. If you require a solution of hydriodic acid, the gas is led through an inverted funnel into a small quantity of cold water. This solution if dilute can be concentrated by distillation, bp 127°C.
Anhydrous Hydrogen Iodide in Acetic Acid2
The presence of molecular iodine in anhydrous solutions of hydrogen iodide in acetic acid gives rise to unstable impurities during the hydriodination of isolated double bonds. This can be overcome by using aqueous hydriodic acid, as the source of hydrogen iodide, from which the iodine has been removed by washing with a solution of an organic soluble ion exchange resin.
In order to develop a practicable synthesis, a procedure for generating anhydrous hydrogen iodide in acetic acid was required. Procedures using molecular iodine (iodine/tetralin at reflux, iodine, and red phosphorus) or compressed hydrogen iodide all proved to be unacceptable. This was because the procedure either was time-consuming or presented safety, handling, or waste management concerns.
All these procedures had one additional and important shortcoming in the context of the proposed chemistry, namely, that traces of iodine, residual during the preparation of the hydrogen iodide, were not readily removable from the resulting solutions produced by passing the gas stream into glacial acetic acid.
The answer was to use analytical grade aqueous hydriodic acid as a readily available and cost effective source of hydrogen iodide. Hydriodic acid of accurately determined concentration was utilised, and all operations were carried out under an argon atmosphere. Traces of molecular iodine were removed by washing with a toluene solution of LA-2 ion exchange resin to produce a colourless and stable aqueous solution. The concentration of hydriodic acid was not affected by the washing process nor was its specific gravity, both of which needed to be accurately determined for the calculation of stoichiometric quantities. The anhydrous acetic acid solutions were prepared by adding the aqueous hydriodic acid to the appropriate quantity of degassed acetic anhydride, with control of the exotherm to below 55°C. The clear and colourless solution was then cooled to 20°C prior to the addition of a solution of alkene in glacial acetic acid. After completion of the required reaction period, the colourless reaction mixture was worked up by vacuum codistillation removal, using toluene, of the majority of the organic and inorganic acids, the product finally being extracted into toluene.
Procedure:
Into an argon-purged separation vessel fitted with a mechanical stirrer is placed hydriodic acid (2165 mL, specific gravity 1.91, 65.0% w/w). A solution of Amberlite LA-2 (0.395 kg) in toluene (5.0 L) is then added to the vessel, and the agitator is used to mix the layers for 2 min. After the layers are allowed to separate, the colourless hydriodic acid layer is run into an argon-purged holding vessel prior to returning to the separator for a single wash with a quantity of degassed toluene. For solutions heavily contaminated with molecular iodine, a second wash with the LA-2 resin solution is required.
Into an argon-purged reaction vessel is then placed acetic anhydride (6.94 L, 99.7%, 73.33 mol) which is vacuum degassed. Washed hydriodic acid (1.973 L, 19.15 mol of HI, 73.33 mol of H2O) is added to the mechanically stirred solution at such a rate that the temperature is maintained below 55°C by the use of external water cooling. If the temperature is allowed to rise above this limit, there is some loss of water vapour by entrainment, and this results in incomplete hydrolysis of the acetic anhydride.
The mixture is stirred for a further 60 min after completion of the addition of the aqueous acid and is then cooled to 20°C prior to the addition of a vacuum-degassed solution of alkene (4.822 mol) in glacial acetic acid (2.0 L) over a period of 10 min.
After completion of the addition, the mixture is stirred for a further 16 h prior to removal of the majority of the acetic acid by vacuum codistillation with 10 volumes of toluene (50 mmHg, <50°C). The dark residue is dissolved in toluene (14.0 L) and then transferred to a separating vessel followed by washing with a 5% solution of sodium thiosulphate (2.0 L) and then deionised water. The thiosulphate wash is first back-washed with a small quantity of toluene, which is combined with the main solution of product.
The organic solution is dried over magnesium sulphate and filtered through a short bed of 100-200 mesh Florisil prior to removal of the toluene under reduced pressure, to leave the product iodoalkene as a colourless to very pale yellow oil. Yield range: 90-97%.
Hydrogen Iodide from Iodine and Tetralin3
Flaky solid iodine of 40 g was dissolved in tetrahydronaphthalene (Tetralin) of 160 g charged in a flask of 500 ml at 40°C to prepare a tetrahydronaphthalene solution of iodine. A flask of 500 ml was charged with tetrahydronaphthalene of 40 g and heated to 200°C. while stirring. The iodine solution prepared above was continuously added thereto over a period of 2 hours while maintaining the above temperature to react them. Crude hydrogen iodide gas generated as the reaction went on was introduced into an aqueous solution of 1 liter to absorb the whole amount thereof. A weight change in this aqueous solution was measured with the lapse of time, and the end point of the first reaction was set at the point where the change thereof was not observed. The yield of the crude hydrogen iodide was 94.6%, and the purity thereof was 99.5% or more.
Anhydrous Hydrogen Iodide, free from Iodine and Phosphonium Iodide4
Summary
The description of a method and apparatus is given for the rapid formation of pure anhydrous hydrogen iodide. This is generated by dropping concentrated hydriodic acid upon phosphorus pentoxide and purified by bubbling through saturated calcium iodide, drying with phosphorus pentoxide and cooling to -30°C.
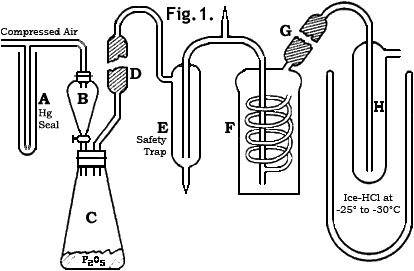
Approximately 150g of conc. hydriodic acid (57% w/w, d. 1.7) was placed in a dropping funnel, B (see figure) and was allowed to drip upon 200 g of phosphorus pentoxide (P2O5) contained in a 500 mL conical flask, C, closed by a wired-on rubber stopper. A slight air pressure insured a steady flow of the acid into the generating flask. The evolved hydrogen iodide, containing traces of iodine, water and phosphine, was purified by passage through an all-glass apparatus. A glass tube, D, filled with phosphorus pentoxide removed mist produced by the vigorous reaction in the generating flask. Iodine and a part of the phosphonium iodide were taken out by a saturated solution of calcium iodide* at 0°C contained in the bubbler, F. Water was absorbed by phosphorus pentoxide in the tube, G, and small amounts of phosphonium iodide were removed by passage through H, maintained at -25°C to -30°C by a cooling bath of ice and concentrated hydrochloric acid. The purified hydrogen iodide may be used as desired.
* The calcium iodide solution, prepared by dissolving 200g of the salt in 100mL of water, removed iodine completely. However, since anhydrous hydrogen iodide in the gaseous state and in solution, for example, in chloroform, is easily oxidized by air and rapidly decomposes when exposed to light, it was necessary to fill the apparatus with nitrogen for the first run and to protect it from light. This latter precaution is of vital importance for obtaining an iodine-free product because even weak, diffused daylight brings about decomposition. Between runs, in spite of these precautions, traces of iodine developed and for this reason it was necessary to discard the gas evolved at the beginning of each run. This formation of free iodine may have resulted in part from the presence of some air introduced while the phosphorus pentoxide supply was being replenished, even though the precaution was taken of passing a rapid stream of nitrogen through the flask before it was connected to the apparatus. Not more than 2g of phosphonium iodide crystallized out during the preparation of 650g of anhydrous hydrogen iodide. Its solubility in the calcium iodide solution is practically nil since the amount at the end of successive runs increased in proportion to the total hydrogen iodide produced. During each run there was formed in the generating flask a small amount of reddish powder, an intermediate reduction product of the phosphorus pentoxide, which was not investigated.
Anhydrous Hydrogen Iodide from KI & Pyrophosphoric Acid5

Figure 1.
Apparatus for the preparation of hydrogen iodide A, 125-ml flask;
B, C, D, intermediate collection tubes of 22-mm tubing; E, storage tube
The apparatus shown in Figure 1 is assembled. The joints should be lubricated with Apiezon (James G. Biddle Co.) or Fluorocarbon grease or pyrophosphoric acid. Silicone grease is less satisfactory.
Potassium iodide, 10g, and 20 mL of pyrophosphoric acid (from 100 mL of 85% phosphoric acid with 100g of phosphorus pentoxide heated to effect solution and then cooled, or polyphosphoric acid diluted to H3PO3 + ~15% P2O5) are added to flask A. A dry ice/trichloroethylene bath is placed about B, and A is heated distilling the hydrogen iodide into B. It is then redistilled into C and D for purification from the small amounts of iodine and phosphorus compounds which are formed. Finally, the HI is distilled into E and is preserved at dry ice temperature.
To dispense the hydrogen iodide, it is warmed to -51°C, its melting point. A syringe needle (preferably of glass) is injected into the liquid hydrogen iodide. As the liquid is brought slowly into the syringe, it vaporizes, and the syringe fills with hydrogen iodide gas. The excess liquid is drained back into the container, and the measured volume of hydrogen iodide gas is transferred to the solution to be analyzed. The solubility of the hydrogen iodide is such that it may be introduced above the surface of the solvent.
Anhydrous Hydrogen Iodide6
C10H12 + 2 I2 -> C10H8 + 4HI
Gaseous anhydrous hydrogen iodide can be prepared by the catalytic union of the elements7,8 and by the reaction of solid iodine with boiling tetrahydronaphthalene.9 The apparatus required for the direct combination of the elements requires considerable time to fabricate and, although the alternative synthesis requires no special apparatus, difficulty is often experienced in controlling the rate of gas evolution. The synthesis described below is a modification of the second method, with the advantage that a steady uniform stream of hydrogen iodide can be generated. Based on the conversion of all the iodine to the hydrogen halide, the yield of hydrogen iodide obtained by this method is nearly quantitative. This is in contrast to the yield of hydrogen bromide by an analogous method,10 which results in conversion of only one-half of the bromine to hydrogen bromide.
Procedure
A 500-mL three-necked ground-joint flask nested in a heating mantle is connected to a reflux condenser, to an inlet gas-delivery tube extending to about 1" from the flask bottom, and to a dropping funnel fitted with a Teflon stopper. Two cold traps fitted with stopcocks are assembled in series to the exit of the reflux condenser through ground-glass joints. The exit from the last trap is connected to a mercury flow bubbler. All joints are lightly coated with Halocarbon grease. The trap connected directly to the reflux condenser is immersed in an ice bath to limit entrainment; the succeeding trap, cooled by liquid nitrogen, is used to collect the anhydrous hydrogen iodide.
The reaction flask is charged with 50 mL of tetrahydronaphthalene and the hydrocarbon is slowly brought to the boiling point, while a solution of 2.56 g (0.01 mol) of iodine in 100 mL of the same hydrocarbon is made and transferred to the dropping funnel. The hydrogen iodide is generated by dropwise addition of the iodine solution to the moderately boiling tetrahydronaphthalene; the rate of addition is sufficient to maintain a slight iodine coloration in the reaction flask. To sweep the hydrogen iodide from the reaction flask, a stream of dry nitrogen is passed into the flask and through the train during the entire reaction. After the addition of the entire iodine charge, the contents of the flask are boiled for a few minutes to remove the residual iodine coloration. The usual vacuum-line techniques11 are used to transfer the hydrogen iodide from the train by distilling through a trap maintained at 0°C and into a storage bulb. The yield of hydrogen iodide is 90%, based on the quantity of iodine employed.
Analysis
The exit gas from the generator ice trap is dissolved directly in water. The hydrogen-ion concentration of the resulting solution is determined by titration with standard base, and the iodide-ion concentration by the precipitation of silver iodide. Only traces of tetrahydronaphthalene are detected by the very slight carbon-hydrogen bond-infrared absorption in the samples of the anhydrous gas prepared by this procedure.