Abstract
The seizure in 1986 of a large-scale clandestine laboratory producing both the N-ethyl and the N,N-dimethyl analogs of 3,4-methylenedioxyphenylisopropylamine (3,4-methylenedioxyamphetamine (MDA) and the recent identification of N-hydroxy MDA hydrochloride (HCl) indicate an interest by illegal laboratory operators in the synthesis of noncontrolled MDA analogs. Currently. identification of these new analogs may be hampered due to lack of available standards or reference data or both. This potential problem prompted the synthesis of the following N-substituted MDA analogs: N-methyl, N-ethyl, N-propyl, N-isopropyl, N-hydroxy, and N,N-dimethyl-MDA. Each compound was prepared by reductive amination of 3,4-methylenedioxyphenyl-2-propanone (MDP-2-P) using the appropriate amine hydrochloride and sodium cyanoborohydride. In addition. the acetyl derivatives of MDA, N-hydroxy-MDA, and MDP-2-P oxime are reported. Spectral and chromatographic data are presented for each of the compounds synthesized.
3,4-Methylenedioxyphenylisopropylamine (MDA), the parent compound for the analogs examined here, was first reported in 19101. The drug gained popularity in the San Francisco area in 1967 and was placed under Federal control in 1970. In 1972. the Bureau of Narcotics and Dangerous Drugs identified an unknown MDA analog as 3,4-methylenedioxymethamphetamine hydrochloride (MDMA·HCl; N-methyl MDA·HCl). Until a few years ago, N-methyl MDA was infrequently abused. However, in the mid 1980s. this compound enjoyed a resurgence of popularity as a "designer drug" touted as "Ecstasy". Increased abuse of the compound resulted in its temporary scheduling as a controlled substance in July 19852 and final confirmation as a Schedule I drug in March 19883. In February 1986, a clandestine laboratory was seized which was producing both the N-ethyl ("Eve") and N,N-dimethyl analogs of MDA. In September 1986, a compound sold as "Fantasy" was identified as N-hydroxy-MDA·HCl.
The proliferation of these analogs has become a source of concern for the forensic science community. These compounds, as well as analogs based on the structure of other controlled substances, are "controlled substance analogs", which refers to potentially physiologically active derivatives of controlled substances, synthesized in an effort to evade the law. These compounds retain the basic chemical structure of the parent controlled substance but have minor structural alterations giving them a unique chemical identity. These analogs are not delineated by the Federal Controlled Substance Act (CSA)4 as passed in 1970 and therefore are not proscribed by that statute. This loophole in Federal law has been remedied by amending the CSA through passage of the Controlled Substance Analogue Enforcement Act of 19865. Briefly, this enactment defines a controlled substance analog as a compound having a chemical structure substantially similar to a controlled substance in Schedule I or II of the CSA.
In addition, for a compound to be a controlled substance analog, it must meet one of two criteria: the compound must have a stimulant, depressant, or hallucinogenic effect on the central nervous system (CNS) that is substantially similar to. or greater than. a controlled substance in Schedule I or II (CSA) or it must be represented or intended by the seller to have the above attributes. This latter part of the definition will encompass compounds for which CNS activity data are not available but for which there is a putative abuse potential. Under the "Analog Law," compounds which meet the specified criteria and are intended for human consumption are subject to the same penalties as for Schedule I controlled substances (CSA). The human consumption clause is included to permit legitimate researchers to continue to design analogs of controlled substances and to conduct and evaluate animal studies without fear of penalties. Should these new analogs prove effective and safe in such preliminary studies, traditional routes for obtaining approval for human testing may then he followed.
The principle of designing new drugs through molecular modification of existing compounds is an established technique in the pharmaceutical industry. Research aimed at maximizing the desired effect of a parent compound and minimizing the undesirable effects results in the development of new derivatives on a continuing basis. When intended for human consumption, these new drugs are subjected to rigid testing and scrutiny to ensure both effectiveness and safety before marketing.
The history of clandestinely manufactured drugs, and especially those which have achieved notoriety as "designer drugs", is markedly different. Safety of the product has been of minor concern to the clandestine chemist. Overdose deaths related to China White "Heroin"6 (α-methyl fentanyl) and the Parkinson-like state induced from by-product contamination of meperidine analogs7-9 illustrate this point. ln most seized clandestine laboratories, quality control is either of low priority or nonexistent. Simplicity of the synthetic route, availability of the required chemicals, and a profitable market are critical elements for the clandestine laboratory operation.
Rarely do clandestine chemists have the ability to devise novel syntheses. As a consequence, the preparation of controlled substance analogs results primarily from substitution of a single reactant in an established method. Often the synthesis of the targeted analog has already been described in detail in the scientific literature. An exact description of the synthesis procedure is usually obtained through a library search of the scientific literature or through purchase of underground drug manufacturing booklets. These drug booklets are generally an edited composite of literature techniques and provide one or more synthetic methods for each of several different drugs. In general, the simplest synthetic methods are the ones most commonly used by clandestine laboratory operators. The following method is an excellent example of a facile synthesis obtained from recognized scientific literature and applied to clandestine drug manufacture.
Experimental Procedures
The synthesis procedure used in this investigation is based on the work of Borch et al.10 and elucidated for MDA and some of its analogs by Braun et al.11. The CNS activity of the analogs presented here has also been described by Braun et al.12. In conformance with their terminology, the analogs here are referred to as 3,4-methylenedioxyphenylisopropylamines rather than 3,4-methylenedioxyamphetamines. Shulgin13 has provided a detailed description to the approaches for naming these compounds.
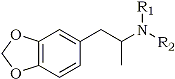
Fig. 1.
Structure of MDA and
N-substituted analogs
Table 1.
Substitutions on MDA for analogs synthesized.
| Chemical Name | Cpd. | R1 |
R2 |
MW |
| 3,4-Methylenedioxy- phenylisopropylamine | 1 | H | H | 179 |
| N-Methyl-3,4-methylenedioxy- phenylisopropylamine | 2 | H | CH3 |
193 |
| N-Ethyl-3,4-methylenedioxy- phenylisopropylamine | 3 | H | C2H5 |
207 |
| N-Propyl-3,4-methylenedioxy- phenylisopropylamine | 4 | H | C3H7 |
221 |
| N-Isopropyl-3,4-methylenedioxy- phenylisopropylamine | 5 | H | CH(CH3)2 |
221 |
| N,N-Dimethyl-3,4-methylenedioxy- phenylisopropylamine | 6 | CH3 | CH3 |
207 |
| N-Hydroxy-3,4-methylenedioxy- phenylisopropylamine | 7 | H | OH | 195 |
Compounds 2 to 7 (Fig. 1, Table 1) were prepared by mixing 3,4-methylenedioxy- phenyl-2-propanone (MDP-2-P) with a methanol solution of the appropriate amine hydrochloride (Fig. 2). Sodium cyanoborohydride was added and the pH of the solution adjusted with hydrochloric acid using methyl red as a visible pH indicator. MDA (Compound 1) was prepared by the same technique but used ammonium acetate as the nitrogen source.
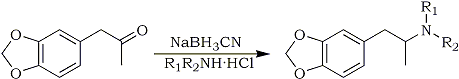
Fig. 2.
Synthetic scheme for N-substituted MDA analogs
In each case the analogs were purified by evaporating the solvent, dissolving the residue in dilute hydrochloric acid and washing with methylene chloride. The aqueous acidic solution containing the analog was made basic with sodium hydroxide, reextracted with methylene chloride, and the solvent evaporated. The crude base which remained was dissolved in isopropyl alcohol and poured into an isopropyl alcohol/hydrochloric acid mixture. Subsequently, ether was added and the precipitated salt collected. Varying amounts of 3,4-methylenedioxyphenyl-2-propanol (MDP-2-Pol) were observed in the reaction mixtures due to reduction of the ketone. The alcohol and any unreacted ketone may be recovered from the methylene chloride washings of the acidic solution described above. Attempts to produce two tertiary amine analogs, N,N-diethyl MDA and N-ethyl,N-methyl MDA, by this procedure were unsuccessful. Application of catalytic hydrogenation for preparation of these two compounds resulted only in trace quantities of the N-ethyl, N-methyl analog sufficient for identification by mass spectrometry. Braun et al.11 were able to prepare the diethyl homolog through reductive alkylation of MDA with acetaldehyde and sodium cyanoborohydride.
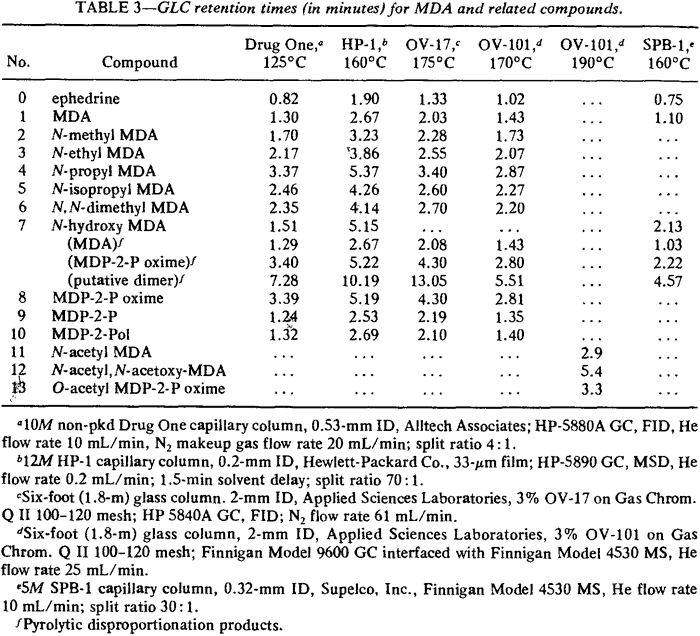
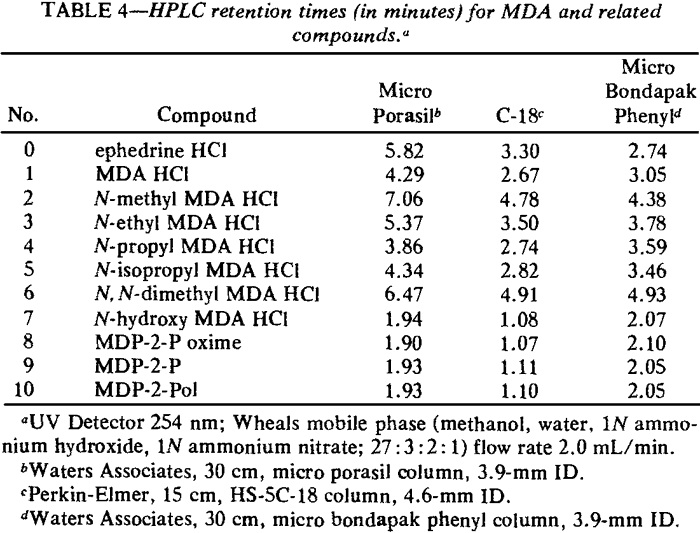
Infrared (IR) spectra were obtained using a Perkin-Elmer Model 1800 Fourier Transform Spectrometer interfaced with a Model 7500 computer. Spectra of the solid compounds listed in Table 2 were prepared using the standard potassium bromide (KBr) matrix technique. For liquids, pure KBr disks were prepared and the liquid coated on the surface of the disk. Gas liquid chromatography (GLC) data were obtained using the instruments. columns, and parameters listed in Table 3. Chloroform solutions (1-3 mg/mL) of each compound were injected into the gas chromatographs (GC) equipped with flame ionization detectors (FID) by automatic sampling devices. GC columns interfaced to mass spectrometers as the detecting device were manually injected with 1-3 mg/mL solutions in methanol. High performance liquid chromatography (HPLC) employed a Perkin-Elmer Series 4 liquid chromatograph with an LCI 100 laboratory computing integrator. Methanol solutions of the compounds were autoinjected by a Perkin-Elmer ISS-100 automatic sampler. A modified Wheals system14,15 was used as the mobile phase for each of the columns listed in Table 4. The ultraviolet (UV) detector was set at 254 µ, a commonly used wavelength in fixed wavelength detectors.
{kind=link}
{kind=link}
Mass spectrometry (MS) employed a Finnigan Model 4530 interfaced to a Model 9600 GC and a Data Control Nova 4 computer. Both electron impact (EI) and chemical ionization (CI/methane) spectra were obtained. EI mass spectra acquired from a Hewlett-Packard Model 5970 mass selective detector (MSD) were essentially the same as those provided by the Finnigan Unit. Proton nuclear magnetic resonance (PNMR) spectra of the title compounds were recorded as the base using a Varian Gemini 300 MHz Fourier Transform NMR. Samples were dissolved in deuteriochloroform, which also served as the reference.
Results and Discussion
Because of the uniqueness of N-hydroxy-MDA among the analogs presented here, a separate discussion of this compound is warranted. N-Hydroxy-MDA has proven difficult to identify by gas chromatography/mass spectrometry (GC/MS) or to screen using GLC because of its tendency to undergo pyrolytic disproportionation (Fig. 3). This chemical process is initiated by heat and results in one molecule of N-hydroxy-MDA oxidizing to 3,4-methylenedioxyphenyl-2-propanone-2-oxime (MDP-2-P oxime), with concomitant reduction of a second molecule to MDA. When pure N-hydroxy MDA·HCl is introduced into the high-temperature injection port of the GC or GC/MS, the resulting chromatogram will exhibit at least two peaks. Examination of the corresponding mass spectra will identify these peaks as MDA and MDP-2-P oxime. These data may lead the analyst to postulate that the material at hand is the impure product of a partially successful attempt to produce MDA by reduction of
Purification of "Street" Samples
Solid samples are ground to a powder and dissolved in dilute hydrochloric acid at room temperature. Any insoluble materials are removed by filtration. The aqueous acidic solution is then extracted with chloroform, made basic with sodium hydroxide, and reextracted with chloroform. The chloroform solution of the basic extract is evaporated to yield the analog.
Compounds 1 to 6 (Table 1) are liquids at room temperature, and care should be exercised to prevent volatilization during removal of the extraction solvent. N-hydroxy-MDA, although initially recovered as a liquid, solidifies upon standing at room temperature. The recovered bases may be converted to the hydrochloride salts by dissolution in an isopropanol/hydrochloric acid solution and precipitating with ether.
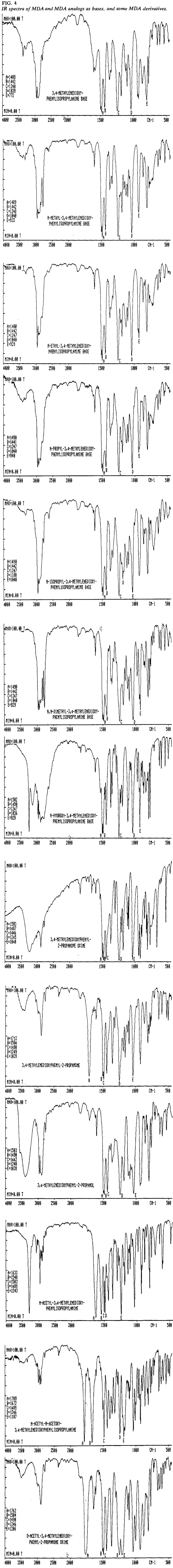
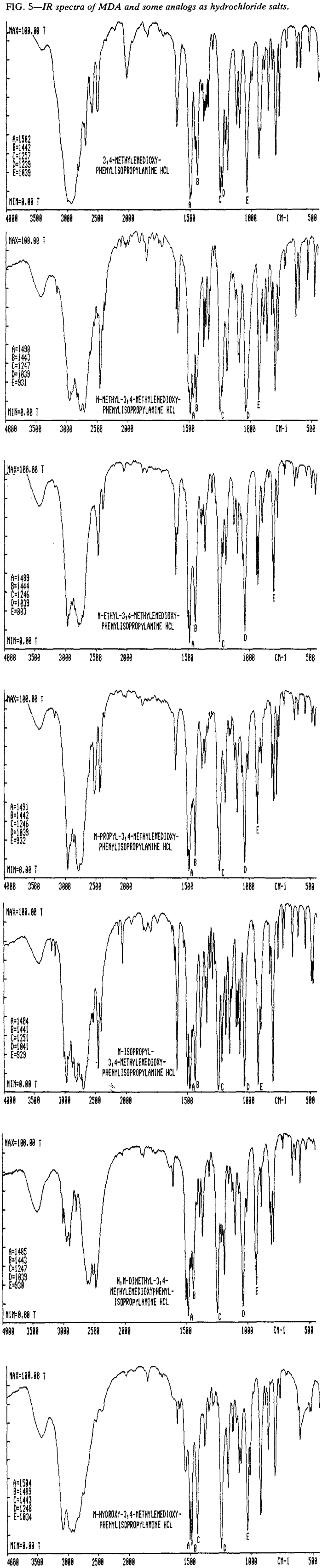
Infrared Spectroscopy
The IR spectra of MDA, its analogs and some related compounds (Table 2) are provided both as the free base (Fig. 4) and, where applicable, as the hydrochloride salt (Fig. 5). With the possible exception of N-ethyl-MDA and N-propyl-MDA, the IR spectra of the analog bases can he differentiated. The hydrochloride spectra of these two compounds show sufficient difference to make identification by comparison of the salt forms possible. It should be pointed out that polymorphism has been encountered in a number of these hydrochloride salts and that five distinct polymorphs have been identified for MDMA·HCl13.
{kind=link}
{kind=link}
Table 2.
List of IR spectra for MDA and related compounds.
No. |
Compound Name |
1 | 3,4-methylenedioxyphenylisopropylamine base (MDA) |
1a | 3,4-methylenedioxyphenylisopropylamine HCl |
2 | N-methyl-3,4-methylenedioxyphenylisopropylamine base (N-methyl MDA) |
2a | N-methyl-3,4-methylenedioxyphenylisopropylamine HCl |
3 | N-ethyl-3,4-methylenedioxyphenylisopropylamine base (N-ethyl MDA) |
3a | N-ethyl-3,4-methylenedioxyphenylisopropylamine HCl |
4 | N-propyl-3,4-methylenedioxyphenylisopropylamine base (N-propyl MDA) |
4a | N-propyl-3,4-methylenedioxyphenylisopropylamine HCl |
5 | N-isopropyl-3,4-methylenedioxyphenylisopropylamine base (N-isopropyl MDA) |
5a | N-isopropyl-3,4-methylenedioxyphenylisopropylamine HCl |
6 | N,N-dimethyl-3,4-methylenedioxyphenylisopropylamine base (N,N-dimethyl MDA) |
6a | N,N-dimethyl-3,4-methylenedioxyphenylisopropylamine HCl |
7 | N-hydroxy-3,4-methylenedioxyphenylisopropylamine base (N-hydroxy MDA) |
7a | N-hydroxy-3,4-methylenedioxyphenylisopropylamine HCl |
8 | 3,4-methylenedioxyphenyl-2-propanone-2-oxime (MDP-2-P oxime) |
9 | 3,4-methylenedioxyphenyl-2-propanone (MDP-2-P/piperonyl acetone) |
10 | 3,4-methylenedioxyphenyl-2-propanol (MDP-2-Pol) |
11 | N-acetyl-3,4-methylenedioxyphenylisopropylamine |
12 | N-acetyl-N-acetoxy-3,4-methylenedioxyphenylisopropylamine |
13 | O-acetyl-3,4-methylenedioxyphenyl-2-propanone-2-oxime |
Gas-Liquid Chromatography
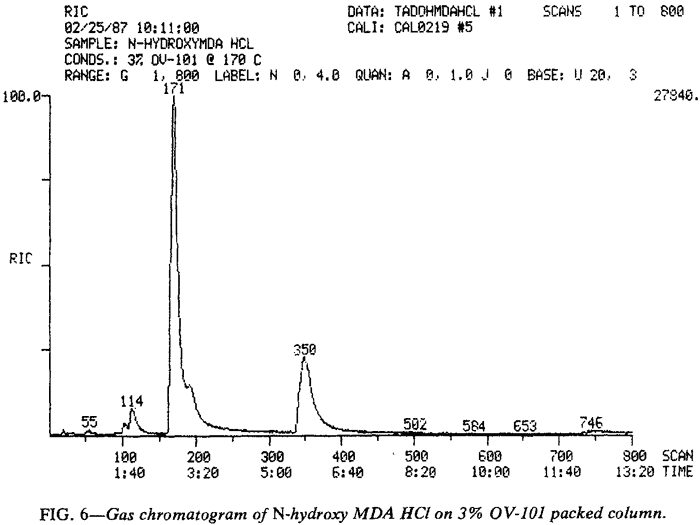
Retention times are listed in Table 3 for several different packed and capillary columns. Ephedrine is included as a reference. Repeated injections of MDA or its analogs on the evaluated packed columns eventually resulted in degradation of peak symmetry and alteration of observed retention times, but this phenomenon can be avoided by using a temperature program. The compound is permitted to elute isothermally (see Table 3). and then the oven temperature is rapidly raised (15�C/min) to 260�C for 2 min before it is returned to the initial temperature. This approach will permit GC quantitation of all the analogs except N-hydroxy-MDA. As discussed earlier, N-hydroxy MDA undergoes pyrolytic disproportionation in the GC injector port (Fig. 3).

Fig. 3.
Pyrolytic disproportionation of N-hydroxy MDA:
Two molecules of N-hydroxy MDA yield one molecule of MDA and one molecule of MDA oxime.
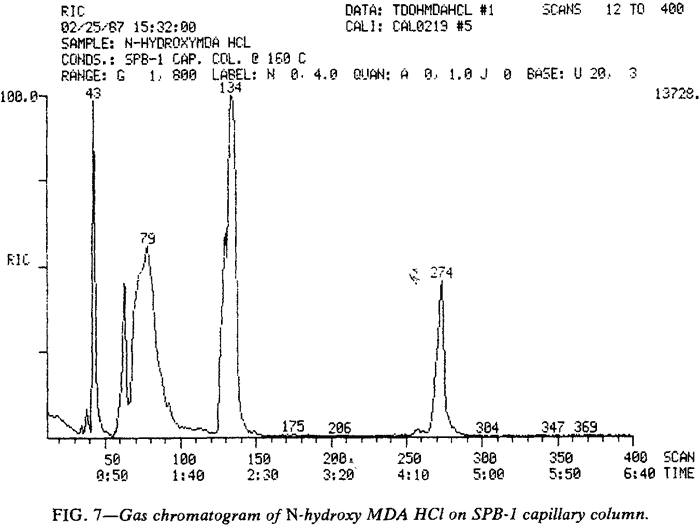
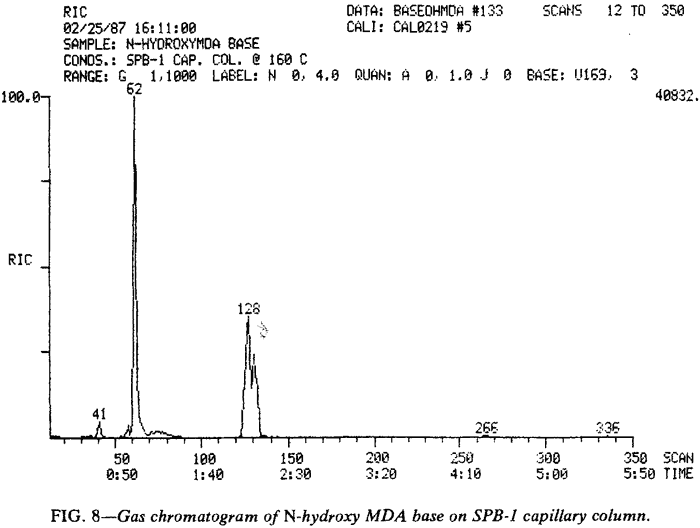
Although MDA (171 s) and MDP-2-P oxime (350 s) provide the primary peaks observed in gas chromatograms of N-hydroxy MDA·HCl when using packed columns (Fig. 6), GCs equipped with capillary columns may provide chromatograms with a different appearance (Fig. 7). The additional peaks are believed to he artifacts of the injection port arising from interaction of the various species available during volatilization, pyrolysis, and disproportionation. The gas chromatographic peak at 4.57 min (274 s) may be a dimer of N-hydroxy-MDA (or MDA or both). MDA dimers have previously been reported16. Injection of N-hydroxy-MDA base (Fig. 8) under identical conditions to those used in Fig. 7 illustrates the effect of hydrogen chloride on the species in the injection port. This comparison also indicates that the additional peaks observed when using the capillary column (Fig. 7) do not arise from the increased resolving power of this type of column. If such were the case, the same peaks would be evident in both Fig. 7 and Fig. 8, but not in Fig. 6. In Fig. 7, N-hydroxy-MDA can be identified as the preceding shoulder (unlabeled at 129 s) on the MDP-2-P oxime peak occurring at 134 s. The broad, unsymmetrical peak at 79 s is, at least in part, MDA. In Fig. 8 , a significant proportion of N-hydroxy-MDA base (128 s) has escaped disproportion into MDA (62 s) and MDP-2-P oxime (unlabeled at 131 s). The EI-GC/MS of these three compounds are presented in Fig. 9.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Unfortunately, packed columns provided essentially identical chromatograms (Fig. 6) whether the base or hydrochloride was injected. and no evidence of N-hydroxy-MDA could be found by GC/MS when packed columns were employed.
High-Pressure Liquid Chromatography
Table 4 provides the data for three types of column packings. In each case, modified Wheals14,15 (methanol, water, 1N ammonium hydroxide, 1N ammonium nitrate; 27:3:2:1) was employed as the mobile phase. HPLC is a suitable method for quantitation. Peak degradation does not occur with subsequent injections as it does with packed GC columns. In addition, UV sensitivity16, even at 254 nm, in quite pronounced (MDA·HCl max/ethanol: λ = 237 nm, ε = 3410; λ = 287 nm, ε = 3580).
Mass Spectrometry
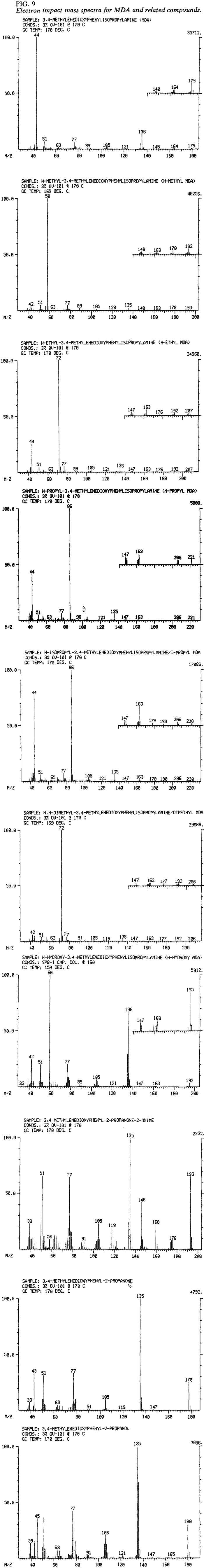
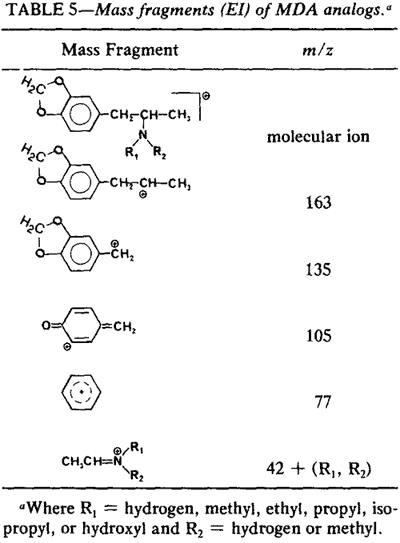
Mass spectra for MDA, the MDA analogs, and several MDA related compounds were obtained as EI spectra (Fig. 9) and chemical ionization (CI) spectra (Fig. 10). The EI mass spectra for MDA and each of the N-substituted derivatives exhibit weak to extremely weak molecular ions which, in some cases, may not be evident without computer enhancement. Insets in the EI mass spectra (Fig. 9) illustrate an increase of intensity of 10 for mass units of 140 and higher. The base peak in each instance results from alpha cleavage and consists of the fragment CH3-CH=N+R1R2 where R1 and R2 represent the appropriate substitutions on the nitrogen atom for that particular analog. Table 5, derived from Lukaszewski16, shows the major EI mass fragments.
{kind=link}
{kind=link}
The difficulties encountered with identification of N-hydroxy MDA were previously discussed. To obtain a suitable mass spectrum of N-hydroxy MDA via GC, it is necessary to introduce the compound as the base using a capillary column. This approach minimizes disproportionation and enables mass spectra of unaltered N-hydroxy MDA (along with MDA and MDP-2-P oxime) to he obtained. Alternatively. a solid probe accessory may be used to obtain a "clean" mass spectrum. Mass spectrometer systems having only packed column capabilities may identify N-hydroxy MDA as its acetyl derivative. The acetyl derivatives for MDA (N-acetyl MDA, MW 221). MDP-2-P oxime (O-acetyl MDP-2-P oxime, MW 235), and N-hydroxy-MDA (N-acetyl-N-acetoxy-MDA, MW 279) were prepared by dissolving MDA·HCl, MDP-2-P oxime and N-hydroxy-MDA·HCl in acetic anhydride and heating on a steam bath until the acetic anhydride evaporated.
Although all three acetyl compounds were initially recovered as liquids, the two amine derivatives solidified upon standing. Each of the acetyl compounds exhibits the expected molecular ion and provides a mass spectrum suitable for confirmation of identity (Fig. 11).
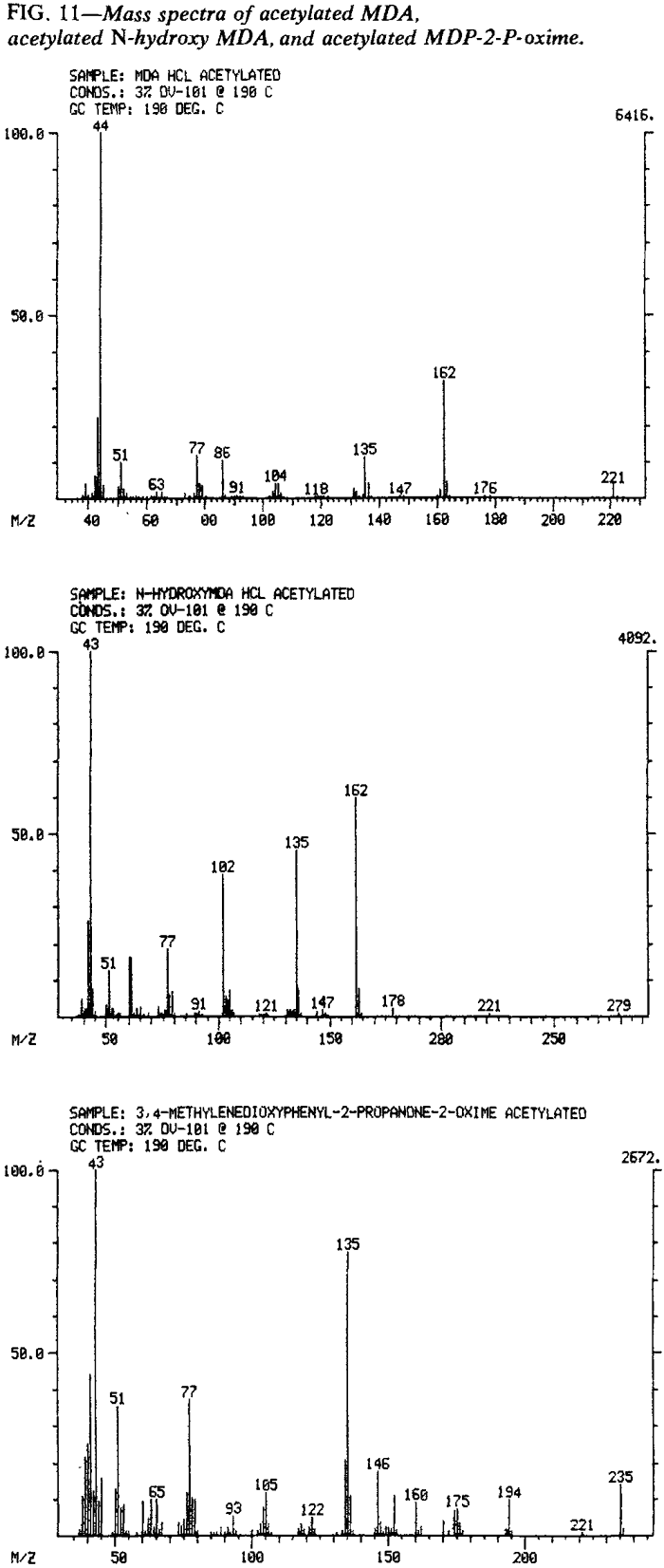{kind=link}
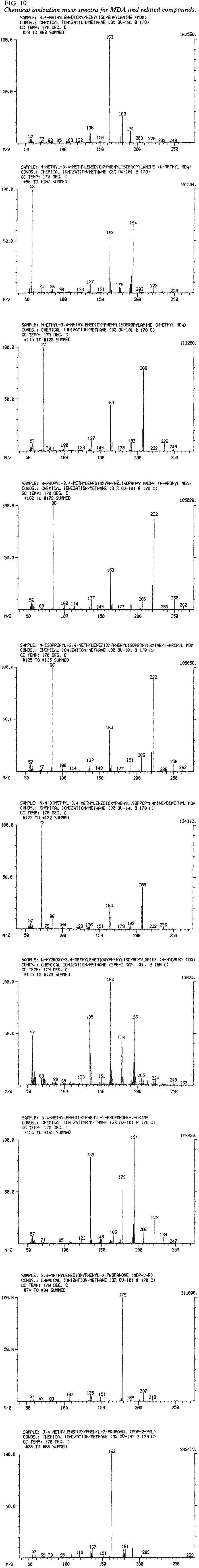
Compounds 1 to 10 (Table 2) were also subjected to CI-GC/MS (Fig. 9). Base peaks for compounds 2 to 6 remain the same as for the EI discussion. Because masses were recorded from 50 to 270 AMU, the expected base peak of 44 AMU for MDA is not reported as such in Table 6. Normalization of the mass spectrum of MDA (Fig. 10) resulted in m/z 163 registering as the base peak. Table 6 indicates the expected CI fragments represented by M (the molecular weight), M+1, M+29, and M+4117,18. Each of the analogs also has fragments at m/z 163, 191, and 203 arising from m + 1, m + 29, m + 41 where m = M - NR1R2 (R1R2 defined in Table 5).
{kind=link}
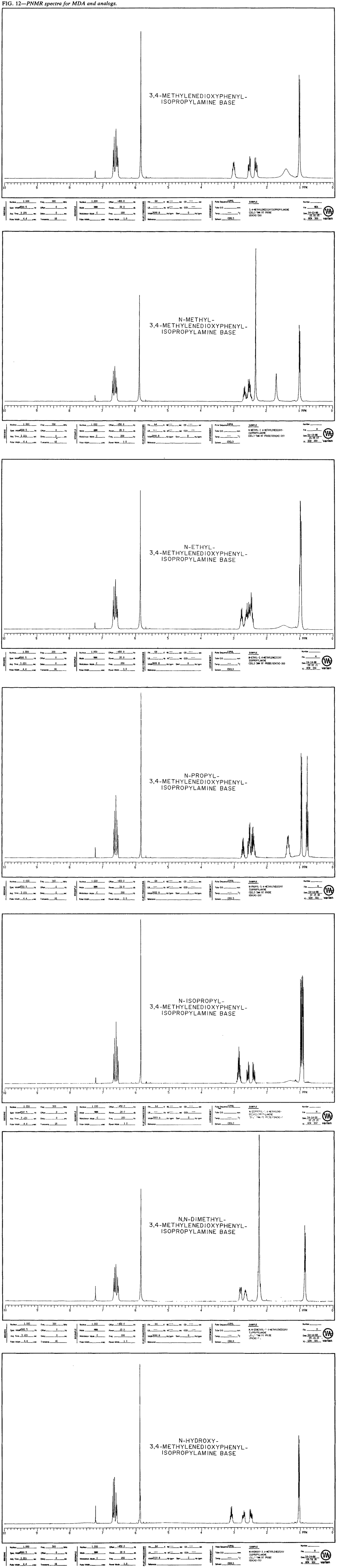
Proton Nuclear Magnetic Resonance
MDA and its N-substituted analogs can easily, and unequivocally, be differentiated by NMR spectroscopy (Fig. 12). The lower field resonances at approximately 6.5 to 6.8 and 5.8 to 5.9 ppm remain essentially the same from analog to analog and result respectively from benzenoid and methylenedioxy protons. The terminal methyl group produces a doublet between 0.8 and 1.1 ppm and is easily recognized except in the isopropyl analog. Careful inspection of the complex pattern overlapping the 0.8- to 1.1-ppm range will reveal the requisite doublet. Distinguishing characteristics are noted in the 2.2 to 3.2-ppm range, with additional resonance interactions evident between 0.8 and 1.5 ppm for the ethyl, propyl, and isopropyl analogs.
{kind=link}
Table 7.
Melting points of salts and derivatives
No. | Compound | Melting Point |
1a |
MDA·HCl | 185-186°C |
2a |
N-Methyl MDA·HCl | 150-152°C |
3a |
N-Ethyl MDA·HCl | 198.5-200°C |
4a |
N-Propyl MDA·HCl | 191.5-192.5°C |
5a |
N-Isopropyl MDA·HCl | 183.5-185°C |
6a |
N,N-Dimethyl MDA·HCl | 169-170°C |
7a |
N-Hydroxy MDA·HCl | 149.5-151°C |
8 |
MDP-2-P Oxime | 86-88°C |
9 | MDP-2-P | |
10 | MDP-2-Pol | |
11 |
N-Acetyl MDA | 91.5-93.0°C |
12 |
N-Acetyl-N-Acetoxy MDA | 72.5-74.5°C |
13 |
O-Acetyl MDP-2-P Oxime |
- Liquid at rt, bp 120-122�C/0.1mm (Fluka)
- Liquid at rt, bp 113-116�C/3mm.
- Liquid at rt, bp 154-157�C/3mm.
Melting Points
Melting points (Table 7) were recorded using capillary tubes in a Thomas-Hoover "Unimelt" melting point apparatus. MDA·HCl and its analogs were purified as described in the experimental procedure. The oxime and the acetylated compounds were purified by dissolving in chloroform and subsequently washing three times with saturated sodium carbonate solution. This was followed by three washings with hydrochloric acid and evaporation of the chloroform. The impure residues were dissolved using large quantities of boiling hexanes and, with the exception of the acetylated oxime, were recovered from the cooled solutions by vacuum filtration. O-Acetyl MDP-2-P oxime is a liquid at room temperature and was further purified by vacuum distillation (Table 7).
Conclusion
The analytical data on MDA, six of its N-substituted analogs, and a number of related compounds will enable the analyst to conclusively identify these substances. Analysts using IR, the "workhorse" for identification in many forensic laboratories, should he aware of the polymorphism exhibited by the hydrochloride salts of MDA analogs. Examination of the analogs as the free base, or in solution, eliminates this potential problem. Alternatively, IR identification of the hydrochloride salt in conjunction with GLC or HPLC retention times will permit the analyst to identify correctly the analog at hand. The PNMR spectra of the analogs are distinctive and will permit the rapid identification of these compounds. EI-GC/MS is less useful for the identification of MDA and the prepared analogs than it is for most classes of illicit drugs. The molecular ions have weak to extremely weak intensities. EI-GC/MS identification should he supplemented with GLC or HPLC retention times. CI-GC/MS results in mass spectra having substantial M+1 fragments.
N-hydroxy MDA is unique among the studied analogs due to its tendency to undergo pyrolytic disproportionation. GLC and GC/MS identification requires injection of the analog as the base onto capillary columns to minimize disproportionation. Alternatives to the use of capillary columns for GC/MS identification are the solid probe technique or conversion of the analog to N-acetyl, N-acetoxy MDA with acetic anhydride.