Organic Syntheses, Coll. Vol. 4, p.573 (1963); Vol. 35, p.74 (1955).
Checked by M. Tishler and H. L. Slates.
1. Procedure
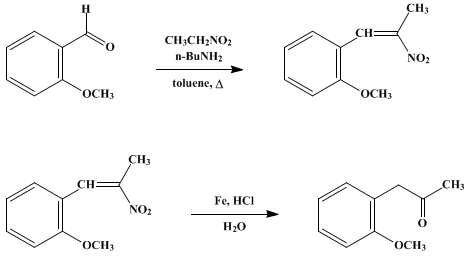
B.
o-Methoxyphenylacetone. A
3-l.
three-necked round-bottomed flask is equipped with an electric heating
mantle, two reflux condensers, a dropping funnel, and a high-speed whip
stirrer (Note 7). The
toluene solution from Part A is placed in the flask, and 500 ml. of water,
200 g. of powdered iron (Note 8), and
4 g. of ferric chloride are added. With vigorous agitation the suspension is heated to about 75°, and
360 ml. of concentrated hydrochloric acid is added over a 2-hour period
(Note 9); heating and stirring are continued for an additional 30 minutes.
The suspension is transferred to a
5-l. three-necked round-bottomed flask and subjected to steam distillation until 7–10 l. are collected
(Note 10). The
toluene layer is removed, and the aqueous layer is extracted with
1 l. of fresh toluene. The combined
toluene layers are agitated for 30 minutes with a solution of
26 g. of sodium bisulfite in 500 ml. of water
(Note 11). The
toluene
layer is washed with water, and the solvent is removed at water-pump
pressure on the steam bath. The resulting orange liquid weighs
107–120 g.. (
65–73%);
nD20 1.5250–1.5270, and is sufficiently pure for most uses. It is purified by distillation through a
12-in. Vigreux column, and the fraction boiling at
128–130°/14 mm. is collected. The yield is
102–117 g. (
63–71%, based on the
methoxybenzaldehyde used),
nD20 1.5250–1.5260
(Note 12),
(Note 13).
2. Notes
2.
o-Methoxybenzaldehyde is available from Eastman Kodak Company, Rochester, New York, and from the Matheson Company, East Rutherford, New Jersey. It may also be prepared according to Baeyer and Villiger.
23.
The
nitroethane was obtained from Commercial Solvents Corporation, Terre Haute, Indiana, as a 90% pure product. The amount used is a 10% excess based on its
nitroethane content. The chief contaminant is
2-nitropropane, which does not interfere in the reaction.
4.
It is desirable to swirl the flask after each addition to prevent the formation of layers.
5. Half of the water is
collected in about an hour, and the theoretical amount (18 ml.) in
about 5 hours. Water removal usually ceases at about 105% of theory.
Insufficient reflux rate causes incomplete water removal or an unduly
prolonged reaction time.
6.
The pure nitroölefin can be obtained by removing the
toluene on the steam bath at water-pump pressure and recrystallizing the resulting oil from
ethanol or petroleum ether.
With petroleum ether, particularly, the volume should be great enough
that the product remains in solution until the solution temperature is
sufficiently low to prevent oiling out. Addition of seed crystals will
encourage crystallization. Alternatively, the yellow oil may be
distilled at reduced pressure. The nitroölefin boils at
135–138°/1 mm. and crystallizes in the receiver when seeded. The yield is
150–175 g. (
80–90%). The yellow crystals melt at
51–52°
when pure. Although no difficulty has been experienced with this
compound, the usual safety precautions should be observed when
distilling an unsaturated nitro compound. The material is somewhat
lachrymatory and irritates the skin.
7.
Rapid agitation is necessary to keep the
iron in suspension and to mix the two liquid layers.
8. A 40-mesh grade was
used, but material up to 100 mesh has been used successfully. However,
with the finer material the reaction is somewhat more vigorous.
9. The reaction mixture
should reflux vigorously. When the addition time is increased to 6
hours, the yield is not appreciably changed. The iron-acid ratio
appears to be important; however, doubling the amounts of both
ingredients produces no change in yield.
10. The 5-l. flask is
either heated with an electric mantle or placed on a steam bath to
prevent condensation of steam and increase in volume of the suspension.
The steam distillation must be continued beyond the point at which the
distillate becomes clear.
11.
The bisulfite treatment removes any aldehydic material present at this point. Since this ketone is quite inert to
sodium bisulfite, the yield is not lowered by this procedure.
12.
The pure ketone has
nD20 1.5240 and a boiling point of
128–130° /14 mm.,
150° /30 mm. Insufficient removal of
o-methoxybenzaldehyde
will cause the refractive index to be high to the extent of about
0.0003 for each per cent present. The use of distilled nitroölefin
eliminates the aldehyde, but this advantage is offset by the
distillation hazard and slightly lower over-all yields.
13. This procedure is
quite general for other aromatic aldehydes. An excess of bisulfite must
be avoided in the washing step, since many phenyl-substituted acetones
react appreciably with it.
3. Discussion
Copyright © 1921-2007, Organic Syntheses, Inc. All Rights Reserved