Abstract
5-Bromo-1,3-benzodioxole (5) on reaction with n-butyllithium in tetrahydrofuran followed by treatment with epichlorohydrin and subsequent quenching by aq. NaOH furnishes a mixture from which 5-oxiranylmethyl-1,3-benzodioxole (2) (safrole oxide) and 5-bromo-4-oxiranylmethyl-1,3-benzodioxole (10) have been isolated and fully characterised. The formation of 2 and 10 clearly indicates that in the reaction of 5 with n-butyllithium, 3,4-methylenedioxyphenyllithium (7) and 2-bromo-5,6-methylenedioxyphenyllithitum (12) are generated.
Haworth and Richardson1 used safrole oxide (2) for the synthesis of the lignan, dehydroanhydropicropodophyllin. Recently2 it has been shown to be a mutagen; some derivatives prepared from it act as insecticides3 and a few derivatives are useful as synergists4 in insecticidal and acaricidal compositions. It has also been used as substrate in the photomeric assay of epoxide hydratase5.
Safrole oxide (2) is usually prepared5 by reacting safrole (8) with m-chloroperbenzoic acid. However, this route is not suitable for the preparation of optically pure safrole oxide in one step, since epoxidation of prochiral alkenes6 with chiral peroxyacids furnishes optically active epoxides in low optical yields. The chlorohydrin (3) was synthesised by Gilman7 by reacting phenyllithium with epichlorohydrin in better yields as compared to methods employing the corresponding Grignard reagents. The conversion of chlorohydrins to epoxides is known8. We, have therefore, examined the chlorohydrin route since both R as well as S enantiomers of epichlorohydrin are available9 in high optical purity and hence the work can be extended for preparing optically active epoxides.
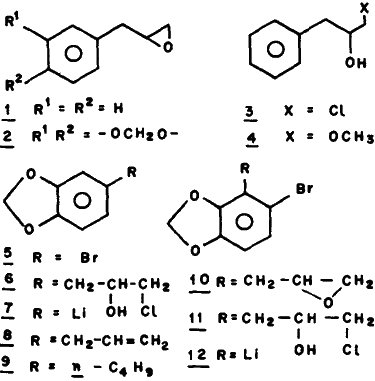
Reaction of phenyllithium with epichlorohydrin followed by quenching with the aqueous NaOH furnished the epoxide (1) in 70% yield; quenching with the methanolic NaOH furnished the methyl ether (4) instead of 1. Formation of 1 provided impetus to carry out a similar reaction of the organolithium compound (7) with epichlorohydrin followed by quenching with aq. NaOH to obtain 2. The bromo compound (5)10 in tetrahydrofuran was reacted with n-butyllithium with the expectation10 that 7 would be formed; the resulting product was reacted with epichlorohydrin in situ and the reaction quenched with aq. NaOH. The product was found to be a mixture (TLC) and it was separated into three main fractions by preparative TLC. PMR of fraction-1 (Rf 0.66) displayed a two proton singlet at δ 5.73 due to methylenedioxy group; the signal at δ 6.43 corresponded to three aromatic H thus suggesting that there is only one substituent on the aromatic ring besides methylenedioxy ring. It was evident from the PMR spectrum that this substituent is n-butyl thus indicating that fraction-1 may be 3,4-methylenedioxy-n-butylbenzene (9). The identity was fully confirmed by direct comparison (IR, PMR and TLC) with an authentic sample11
TLC of fraction-2 showed two spots (Rf 0.46 and 0.43); these components were separated by fractional distillation. The lower boiling component was identified as safrole oxide (2) by direct comparison with an authentic sample. The mass spectrum of the higher boiling component showed molecular ion peaks of equal intensities at m/z 256 and 258 indicating the presence of one bromine atom. This data together with PMR spectrum indicated the higher boiling component to be the oxide 10.
Fraction-3 was again a mixture of two components (Rf 0.26 and 0.22), as revealed by TLC. This fraction showed a strong band at 3300 cm-1 in the IR spectrum suggesting the presence of hydroxyl group. The mass spectrum exhibited peaks at m/z 292, 294 and 296; the relative abundances of ions were consistent with the presence of one bromine and one chlorine atom thus indicating that one of the components was the chlorohydrin (11). The PMR spectrum suggested that fraction-3 was a mixture of the chlorohydrins (6) and (11). This conclusion was supported by the observation that treatment of fraction-3 with aq. alkali furnished a mixture of oxides (2) and (10).
Survey of literature reveals that in certain cases lithiation on an aromatic ring can take place without displacing the bromine atom on the aromatic ring. For example p-bromoanisole is known12 to furnish 5-bromo-2-methoxyphenyllithium on treatment with n-butyllithium. Ether oxygen attached to aromatic ring is known13 to direct lithiation to the ortho position and this reaction is particularly facile in tetrahydrofuran medium. In the bromo compound (5) there are two hydrogens ortho to ether oxygen; our results indicate that the more hindered ortho hydrogen has been abstracted. In the lithiation reactions, there are precedents in literature for the abstraction of sterically crowded hydrogens14,15. The ease of abstraction of hydrogen is dependent on its acidity16,17.
Experimental
1-Methoxy-3-phenyl-2-propanol (4)
To phenyllithium, prepared from lithium metal (0.15 g) and bromobenzene (1.57 g) in anhydrous ether (10ml), was added epichlorohydrin (0.92 g) with stirring at -15°C. The reaction mixture was stirred at 0°C for 0.5 hr and at 25°C for 3 hr. Subsequently a solution of NaOH (0.4 g) in methanol (5 ml) was added and the resulting mass stirred at room temperature for 1 hr. The product was isolated through column chromatography over alumina to furnish 4 (0.91 g).
Lithiation of 3,4-methylenedioxybromobenzene (5) and reaction with epichlorohydrin
Bromocompound (5) was prepared10 from 1,3-benzodioxole and its purity checked through glc. n-hexane solution of n-butyllithium (2.95 N, 2.24 ml) was added to a solution of 5 (1.36 g) in tetrahydrofuran (20 ml) at -78°C. After 5 min epichlorohydrin (0.60 g) was added. The reaction mixture was kept at -78°C for 30 min and the temperature allowed to rise to -10°C. A solution of NaOH (1.6g) in water (16 ml) was added and the reaction mixture stirred at room temperature for 1h. The product isolated after usual work-up was separated into three main fractions through preparative TLC on silica gel. The TLC plates were developed with 85:15 mixture of pet. ether-acetone.
Fraction-1 (0.08 g), Rf 0.66 has been identified as 9 by direct comparison (IR, PMR, TLC) with an authentic sample11.
Fraction-2 (0.48g) showed two spots (Rf 0.46 and 0.43). The lower spot corresponded to safrole oxide (2); areas of PMR signals at 5.88 and 5.95 were nearly equal indicating that fraction-2 was an equimolecular mixture of 2 and 10. Fractional distillation of fraction-2 using a vigreux column furnished (i) a lower boiling cut, bp 120°C/2mmHg, identified as 2 and (ii) a higher boiling cut, bp 150°C/2 mmHg.
Fraction-3 (0.12g) showed two spots (Rf 0.26 and 0.23). Fraction-3 (0.05g) was stirred with a mixture of NaOH (0.4 g) and water (4 ml) at room temp. for 2 hr. The product isolated was a mixture of 2 and 10 as shown by TLC and GLC.