Abstract
Trans 4-hydroxycrotonic acid (T-HCA) has been identified in central nervous system of mammalians as a naturally occurring substance, which may compete with 4-hydroxybutyric acid (GHB) for specific biological targets, such as high affinity binding sites, uptake systems and metabolism enzymes. T-HCA has been tritiated at the 2,3-positions, using a multi-step synthesis and a one-pot reaction for the three last critical steps. Thus, T-HCA-[2,3-3H] was obtained with a specific radioactivity of 45 Ci/mmole (1.66 TBq/mmole) and a radiochemical purity of 97%.
Introduction
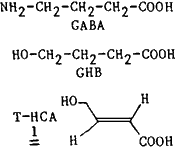
4-Hydroxybutyrate (GHB) is present in the brain of mammalians1 and is formed from 4-aminobutyric acid (GABA)2. This compound has numerous neuropharmacological and neurophysiological properties3,4. The existence of specific high affinity binding sites5,6 and uptake systems7 for GHB sets forth arguments in favour of a role for this substance in central nervous transmission4.
First considered as a synthetic semi-rigid analogue of GHB, trans-4-hydroxycrotonic acid (T-HCA) 1 has been recently identified as a naturally occurring substance in human renal tissue8 and rat brain9. Its interaction with the specific GHB biological targets5,7,10 and its unknown metabolism prompted us to synthesize tritiated T-HCA.
Discussion
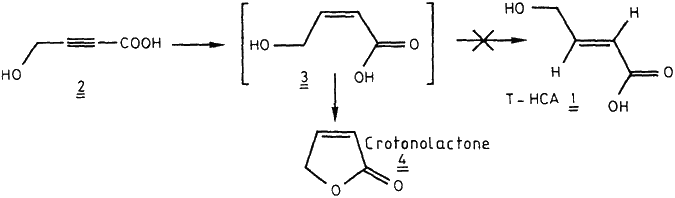
The 4-methylene protons of T-HCA are labile as a result of possible 1,3-sigmatropic rearrangements11-12 into succinic semialdehyde. Thus the 2- and 3-positions were suitable for labelling of T-HCA by tritium. 4-Hydroxytetrolic acid 2 was first considered as a valuable precursor for the preparation of [3H]-T-HCA 1 according to Scheme 1. Semi-hydrogenation of 2 in presence of Rosenmund catalyst13 led to the expected isomer, Z-4-hydroxycrotonic acid 3 which spontaneously lactonized into 4. Hydrogenation of the triethylammonium salt of 2 yielded only side-products as a result of prototropic rearrangements15 as mentioned above. However, complete hydrogenation of 2 afforded [3H]-GHB with high specific radioactivity (100 Ci/mmole, 3.7 TBq/mmole).
Scheme 1
Scheme 2
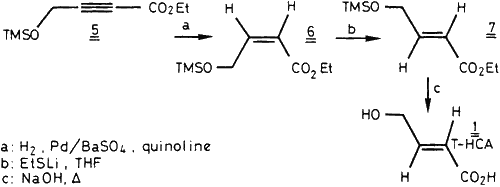
In an alternative route (Scheme 2), the O-TMS derivative 5 of 4-hydroxy- tetrolic acid was partially hydrogenated (Pd/BaSO4) to the Z-ester 6. Isomerization of 6 by lithium ethanethiolate16 gave 7 which hydrolysis provided E-4-hydroxycrotonic acid 1. Drastic anhydrous conditions were required for the isomerisation step and were critical at the mg scale. Tritiation experiments only gave side-products, however, probably Michael adducts16 of ethanethiol.
Scheme 3
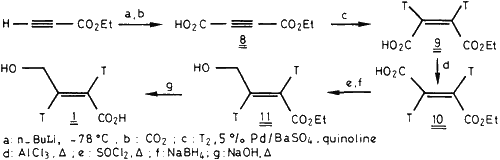
Therefore, acetylene dicarboxylic acid monoethylester was chosen as starting material for 3H-T-HCA preparation (Scheme 3). 8 was obtained by carbonation of metallated ethyl propiolate with carbon dioxide. Partial hydrogenation with tritium gas and Rosenmund catalyst led to maleic acid monoethyl ester [2,3-3H] 9. Cis-trans isomerization of electron-poor acrylic derivatives such as 9 is well documented in the patent literature. Thermal isomerization of 9 using AlCl3 as catalyst17 afforded 10 with a satisfactory yield (36%) after purification by medium pressure liquid chromatography (50% of radioactivity were lost in polymeric materials). Further reduction of 10 with LiBH4 gave a mixture of saturated compounds resulting from 1,4- instead of 1,2-reduction mechanisms. Therefore, 10 was converted to its acyl chloride18 which was reduced by NaBH4 to give 11. Without purification, 11 was hydrolyzed to T-HCA-[2,3-3H] 1. The latter was purified by medium pressure liquid chromatography and preparative thin-layer chromatography.
The chemical and radiochemical purity of tritiated T-HCA 1 were determined by HPLC. Labelling positions and configuration of the final product were checked by 3H-NMR analysis. The specific activity of 3H-HCA 1 was 45 Ci/mmole (1.66 TBq/mmole).
This tritiation procedure is long but is satisfactory as the last three steps are a one-pot reaction. Preliminary binding studies of 3H-T-HCA 1 suggest that high affinity T-HCA binding sites constitute a sub-class of GHB binding sites.
Experimental
4-Hydroxybutyrate-[2,2,3,3-3H4] (3H4-GHB)
4-Hydroxytetrolic acid 2 (5 mg) was tritiated in presence of 10% Pd/C (10 mg) for 1 h to provide 5 Ci (0.185 TBq) of product which was successively purified by paper chromatography (ethanol-water-ammonia, 92:8:1) and thin layer chromatography on silica gel (isopropanol: 75, ammonia: 15, water: 10).
700 mCi (25.9 GBq) of 4-hydroxybutyrate-[2,2,3,3-3H] were obtained with a radiochemical purity over 97% (by thin-layer chromatography on silica gel: isopropanol/ammonia/water: 75/15/10, Rf = 0.3) and by high-performance liquid chromatography on an Aminex HPx87 H column eluted by 0.013 N H2SO4, VR = 12.5 ml).
4-Trimethylsilyloxy-2-butynoic acid ethyl ester 5
4.0 g (0.04 mole) of 4-hydroxytetrolic acid 2 in 50 ml of EtOH was reacted with 1 ml of conc. H2SO4 at room temperature for 24 h. The solution was then carefully neutralized with 1.5 g (0.01 mole) of K2CO3. After evaporation of the solvent, the mixture was poured into water and extracted with ethyl ether. After drying and removal of the solvent, the crude oil was distilled under reduced pressure (78°C, 0.05 mm Hg) giving 3.95 g (83%) of pure ethyl 4-hydroxytetrolate. This ester (0.115 g, 0.9 mmole) was dissolved in 10 ml of anhydrous acetonitrile and the solution and treated with 0.250 g (1 mmole) of bis-trimethylsilyltrifluoroacetamide and 0.120 g (1 mmole) of diisopropylethylamine. The reaction mixture was stirred overnight at room temperature under nitrogen atmosphere. After evaporation of the solvent, the crude product was taken up with ethyl ether, washed with water, and dried over Na2SO4. After removal of the solvent, the solution was distilled using a bulb-to-bulb distillation apparatus (115°C, 0.2 atm.), affording 0.190 g of 5 as a yellow oil (quantitative yield).
(Z)-4-Trimethylsilyloxy-2-butenoic acid ethyl ester 6
0.805 g (4 mmoles) of ester 5 in 15 ml of absolute MeOH was vigorously stirred at room temperature under hydrogen with 25 mg of 5% Pd-BaSO4 catalyst. When about 90 ml of hydrogen were adsorbed, the catalyst was filtered off and the solvent removed under vacuum.
(E)-4-Trimethylsilyloxy-2-butenoic acid ethyl ester 7
To 0.609 g of 6 (3 mmoles) in 10 mL of anhydrous THF at 0°C was added dropwise 1 mmol of lithium ethanethiolate16 and the solution was left at room temperature for 1 h. After evaporation of THF, the crude product was taken up in ethyl acetate and washed with H2O. The organic layer was dried and evaporated affording the crude E isomer 7.
E-4-Hydroxycrotonic acid (T-HCA) 1
0.130 g (1 mmole) of ester 7 in 3 mL of EtOH was reacted at 4°C with 0.5 ml of a 10 N NaOH solution. The mixture was heated at 60°C (external temperature) for 30 min. The solution was then cooled and carefully acidified to pH 1 by 1N HCl. After removal of the solvents under vacuum, the residue was triturated with 10 ml of warm ethyl acetate and the solid filtered off. After removal of the solvent in vacuo, 100 mg (98%) of crude T-HCA 1 (mp 104°C) were obtained and recrystallized in ethyl acetate affording pure T-HCA 1 (bp 108°C; lit. 104°C20).
2-Butynedioic acid monoethyl ester 8
0.980 g (10 mmoles) of ethyl propiolate in 60 mL of anhydrous THF was cooled at -78°C. To the reaction mixture were added dropwise 8.5 mL (12 mmoles) of a 1.4 N BuLi solution over 30 minutes. CO2 gas dried over concentrated H2SO4 was bubbled through the solution at -78°C for 15 min. The medium was hydrolyzed with 50 mL of a NH4Cl saturated aqueous solution. After removal of THF under reduced pressure, the medium was extracted with ethyl ether. The aqueous phase was carefully acidified to pH 1 with dilute H2SO4 and evaporated to dryness. The resulting solid was triturated with three portions of ethyl ether and filtered off.
After evaporation of the organic layer, the remaining solution was distilled under vacuum in a bulb-to-bulb distillation apparatus (bp 100°C/150 mTorr) affording pure 8 (0.950g; 68%).
(Z)-2-Butenedioic acid monoethyl eater [2,3-3H] 9
8 (50 mg, 0.342 mmol) in 2 ml of toluene, 5.1 mg of 5% Pd/BaSO4 catalyst and 100 µL of quinoline were placed in a reaction flask and degassed in vacuo. 50 Ci (1850 GBq) of tritium gas were slowly introduced. After 20 minutes, 8.6 mL of gas were consumed, and the catalyst was filtered off. The solution was then taken up with 3x5 mL portions of MeOH and evaporated in vacuo to remove labile tritium. The crude product was dissolved in 80 mL of water and extracted with 10 mL of toluene. After evaporation, the residue was chromatographed by medium pressure (10 bars) chromatography using a silica column (diam. = 2.5 cm) eluted by hexane/CH2Cl2/AcOH (6:3:1) (flow rate: 2 mL/min). 14 Ci (518 GBq) of 9 were obtained. Radiochemical purity >97% (checked by thin-layer radiochromatography on silica gel with hexane/CH2Cl2/AcOH 5:4:1).
(E)-2-Butenedioic acid monoethyl ester 10
A mixture of 9 (14 Ci, 518 GBq) and anhydrous AlCl3 (1.2 mg) was heated at 65°C in a 3 mL conic flask for 18 h and then added to a mixture of CH2Cl2/EtOH (V/V). After evaporation, 10 (7 Ci, 259 GBq) was obtained with a radiochemical purity of 90% (TLC: hexane/CH2Cl2/AcOH, 5:4:1, Rf = 0.52). 7 Ci of 9 were lost as tars insoluble in organic solvents. The crude 10 was dissolved in 5 mL of EtOH, filtered on a 0.22 µm filter and purified by medium pressure (10 bars) liquid chromatography (silica gel, column diam. 2.5 cm, eluted with hexane/CH2Cl2/AcOH 7:2:1). 5 Ci (185 GBq) of 10 were obtained with a minimum radiochemical purity of 97% (TLC hexane-dichloromethane-acetic acid: 5/4/1).
4-Hydroxy-2-butenoic acid [2,3-3H] ([3H]-T-HCA) 1
5 Ci (185 GBq) of 10 in 3 mL of dry ethyl ether were heated for 70 min in a conic flask in presence of 1 mL of freshly distilled thionyl chloride. The mixture was cooled and concentrated under vacuum (50 mmHg). The cold reaction medium was taken up in 0.2 mL of anhydrous acetonitrile and reacted with NaBH4 (4.2 mg, 0.111 mmol) at 4°C for 20 minutes. The mixture was hydrolyzed with 1 mL of a 1N HCl solution. After removal of the solvent in vacuo, the crude product was dissolved in 2 mL of water and after evaporation 10 mL of EtOH were added. The total amount of radioactivity was 1.6 Ci (59 GBq). Thin-layer chromatography on silica gel (cyclohexane/CH2Cl2/AcOH 5:4:1) allowed the identification of the mixture of the starting ester 10 (18% of total radioactivity, Rf = 0.52), the ester-alcohol 11 (18%, Rf = 0.36) and T-HCA 1 (8%, Rf = 0.10).
A second hydrolysis in the same conditions considerably increased the yield of 1 (26%).
Tritiated T-HCA 1 was isolated by medium pressure (10 bars) liquid chromatography on silica gel (column (diam. 2.5 mm) eluted with a mixture of hexane/CH2Cl2/AcOH 7:2:1. An aliquot of 38 mCi (1.4 GBq) of T-HCA 1 (radiochemical purity 31%) was purified by preparative TLC on silica gel with the same solvents affording 9 mCi (333 mBq) of radiochemically pure [3H]-T-HCA 1. The radiochemical purity was determined by HPLC using an Aminex HP x 87 column and a 0.013N H2SO4 solution as solvent (VR = 9.0 mL)-3H-NMR analysis allowed the identification of three isotopomers: [2-3H], [3-3H] and [2,3-3H2] T-HCA derivatives in 21, 19, and 60% respective yields.