A Study and Identification of Potential By-Products of Sibutramine
G.O. Reddy, M.R. Sarma, B. Chandrasekhar, J. M. Babu, A.S.R. Prasad, and C.M.H. Raju
Org. Proc. Res. Dev. 3, 488-492 (1999)
Abstract
In the synthesis and process development of sibutramine (9), the isolation and characterization of two potential by-products namely heptane dinitriles (4a-b) and bis-cyclobutyl alkylamine (10) have been studied. The key steps in the synthesis of sibutramine which have contributed to the formation of above by-products are cycloalkylation of 4-chlorophenyl acetonitrile (1) and tandem Grignard reduction on 1-(4-chlorophenyl)-cyclobutylcarbonitrile (3).
Novel diacid accelerated borane reducing agent for imines
Zhi-Hui Lu, Nandkumar Bhongle, Xiping Su, Seth Ribe and Chris H. Senanayake
Tetrahedron Letters 43, 8617–8620 (2002)

Abstract
A remarkable effect of diacids in modulating the reactivity of borane has been discovered. This novel
process provides a rapid and excellent access for reduction of a variety of imines with different functionalities.
New resolution approach for large-scale preparation of enantiopure didesmethylsibutramine (DDMS)
Zhengxu Han, Dhileepkumar Krishnamurthy, Q. Kevin Fang, Stephen A. Wald and Chris H. Senanayake
Tetrahedron: Asymmetry 14, 3553–3556 (2003)
Summary
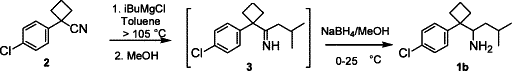
The unoptimized reaction was originally performed by Grignard addition to 1-(4-chlorophenyl)-1-cyclobutylcarbonitrile 2 in toluene at 90–95°C for 18 h, followed by reduction using 3 equiv. of NaBH4 in iso-propanol under refluxing for >20 h, and the reaction was quenched with water to furnish the product.
After a series of experiments an optimum procedure was established, which uses the addition of i-BuMgCl to 2 at >105°C for 2 h, followed by quenching with methanol. The imine intermediate 3 was reduced using one equivalent of NaBH4 at 0–25°C for 1 h. An efficient quenching process was identified by adding the reaction mixture to a 2 M HCl aqueous solution at 0°C, which generated a homogeneous reaction mixture that can be easily stirred. After work-up, this optimized process afforded a crude racemic 1b in toluene with excellent yield (>95%). The optimized experimental conditions proved highly desirable for large-scale production (see below).
Experimental
A 1 L three-necked round-bottomed flask was charged with 1-(4-chlorophenyl)-1-cyclobutyl- carbonitrile (50.0 g, 261 mmol) and toluene (150 mL), followed by iso-butyl magnesium chloride (395 mL, 1.0 M in MTBE), and the resulting mixture was distilled until the internal temperature reached 105°C. After stirring at that temperature for 2 h, the mixture was cooled to 0°C and methanol was added slowly (295 mL), followed by sodium borohydride (10.4 g, 1.06 equiv.) portion-wise. The resulting mixture was stirred at rt for 15 min and was added to a 2N HCl solution (330 mL) slowly, stirred for 15 min and the phases were separated. The aqueous phase was extracted with toluene (300 mL), the combined organic phases were distilled to remove methanol, and then washed with aqueous NaOH solution (1.5 M, 100 mL) and water (100 mL) twice. The resulting organic phase was heated to 50–60°C, followed by an addition of D-tartaric acid (40.0 g) in water (80 mL) and acetone (40 mL) slowly. The reaction mixture was azeotrope distilled until the internal temperature reached 92°C and then cooled to ambient temperature in 1–2 h. The slurry was filtered, and the wet cake was washed with MTBE (100 mL) and dried at 40–45°C under reduced pressure to afford (RS)-DDMS·D-Tartrate (100.5 g) in 95.8% yield.
{kind=link}