Theory of Liquid-Liquid Extraction
As we saw in the previous lab, in chemistry, extraction is the physical process by which a compound (or a mixture of compounds) is transferred from one phase into another. The isolation of trimyristin form nutmeg is an example of a solid-liquid extraction. It is also possible to partition the components of a mixture between two immiscible liquids (i.e., liquids that will not dissolve in each other and form two distinct phases when combined). This process is called a liquid-liquid extraction.
There are two general types of liquid-liquid extractions:
1. an organic solvent extraction, in which an organic solvent with a high affinity for the desired compound is used to extract the compound from another solution, and
2. an acid-base extraction, in which an organic acid or base is extracted from an organic solvent by using an aqueous solution of an inorganic base or acid, respectively. A neutralization occurs which converts the compound into an ionic, water-soluble salt, causing it to transfer from the organic phase to the aqueous phase.
Extraction with organic solvents
Liquid-liquid extractions usually involve water and an organic solvent. Most common organic solvents (diethyl ether, ethyl acetate, toluene, methylene chloride) are immiscible in water. If you place 50 mL of ethyl acetate and 50 mL of water in a flask and stir the solution to mix it, you will not obtain a homogeneous solution. Rather, if the solution is allowed to stand after stirring, two distinct liquid phases will form in the flask: the more dense solvent as the lower layer and the less dense solvent as the upper layer.
Most organic solvents are much less polar than water. A general rule of thumb for solubility states that like dissolves like. Polar compounds are more soluble in polar solvents than in nonpolar solvents, and vice versa. The selective solubility of different compounds in polar versus nonpolar solvents allows the separation of the compounds in a mixture by liquid-liquid extraction.
Suppose
that we add a compound X to a flask containing ethyl acetate and water,
and stir the contents of the flask to mix them. After mixing, the ethyl
acetate and water will separate into two distinct phases, and compound
X will be found dissolved in both the ethyl acetate layer and in the
water layer. How compound X distributes between the two solvents is
based on the solubility of X in each of the two solvents: more of
compound X will be found in the solvent in which it is more soluble.
The ratio of the concentrations of X in each of the immiscible solvents
is called the distribution coefficient or the partition coefficient, Kd, where
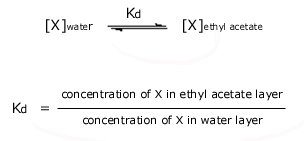
The
value of the distribution coefficient depends on the solubility of the
compound in the two solvents in the system. In the above system, if
compound X has a higher solubility in ethyl acetate than in water, at
equilibrium the concentration of X in ethyl acetate will be greater
than the concentration of compound X in water, and the value of the
distribution coefficient Kd will be greater than 1. If
instead compound X has a higher solubility in water than in ethyl
acetate, at equilibrium the concentration of X in water will be greater
than the concentration of compound X in ethyl acetate, and the value of
the distribution coefficient Kd will be less than 1.
The efficiency of a liquid-liquid extraction depends on the distribution coefficient of the desired compound between the two solvents. If we want to extract an organic compound from an aqueous solution into an organic solvent, it is desirable to use a solvent that has a much higher affinity for the compound than does water. For example, at 25 oC, the solubility of benzoic acid in water is 3.4 g per liter while the solubility of benzoic acid in chloroform (CHCl3) is 222 g per liter. Water and chloroform are immiscible solvents. If a solution of 1 g of benzoic acid in 400 mL of water is extracted with 400 mL of chloroform, we would expect most of the benzoic acid to be transferred to the chloroform layer in which it is more soluble. The benzoic acid will distribute itself between the two solvents in the ratio of the solubilities in each solvent:

No matter how much benzoic acid is present in the system, it will always be distributed between the chloroform and water so that the ratio of the concentration in each solvent is 65.3.
From
this estimate of the distribution coefficient, we can calculate how
much benzoic acid is present in the chloroform and water layers after
the extraction. Let x = grams of benzoic acid in the water layer and y
= grams of benzoic acid in the chloroform layer. Since we started with
1 g of benzoic acid,
x + y = 1 . Using this equation along with the
value for the distribution coefficient calculated above, we can
determine the concentration of benzoic acid in each layer:

or, since the volumes of both solvents used are the same:

The total amount of benzoic acid present is (x + y) = 1. Rearranging this equation and substituting for x in the previous equation gives
Solving this equation for y gives 0.015 g (15 mg) of benzoic acid in the water layer, and, since the total amount of benzoic acid is 1 g, there is 0.985 g (985 mg) of benzoic acid in the chloroform layer.
Multiple extractions
In the previous example, one extraction with 400 mL of chloroform removed 98.5 % of the benzoic acid from the aqueous solution. If we divide the 400 mL of chloroform used in half and do two successive extractions of the aqueous phase, the amount of benzoic acid extracted will increase.
The equation for the distribution coefficient for two 200 mL chloroform extractions of the 400 mL aqueous solution of benzoic acid is

In the first extraction, 1 g of benzoic acid is distributed between the phases, so (x + y) = 1 as before. Solving the two equations in two unknowns gives x = 0.97 (g in CHCl3) and y = 0.03 (g in H2O). When the aqueous phase is extracted a second time with a fresh 200 mL of chloroform, only 0.03 g of benzoic acid is left in the aqueous phase to distribute between the two solvents. In this extraction the equation for the distribution coefficient is the same but (x + y) = 0.03, and solving for x and y, the amount of benzoic acid in each layer after the second extraction gives x = 0.0291 (g in CHCl3) and y = 0.0009 (g in H2O). Combining the amounts of benzoic acid found in the two chloroform extracts gives 99.91% ( 0.9991 g of the original 1 g) of the benzoic acid extracted into the chloroform layer by using two 200 mL extractions instead of 98.5% removed with one 400 mL extraction. In general, it is always more efficient to carry out several extractions using a small volume of solvent each time than to carry out a single extraction using a large volume of solvent.
Acid-base extraction
Organic compounds are classified as being neutral, acidic, or basic depending on the types of functional groups they contain. Many organic compounds, although just slightly polar overall, contain functional groups that can act as a Brønsted-Lowry acid or base (i.e., they can donate or accept a proton, respectively). Carboxylic acids, phenols, and thiols are examples of acidic functional groups; substituted amines (including anilines) are examples of basic functional groups. Although the water-solubility of these compounds is often limited because of their overall nonpolar character, their aqueous solubilities can be dramatically increased through an acid-base neutralization reaction. This changes the compound into an ionic salt that is very water soluble and will distribute almost completely into the aqueous layer.
To illustrate how an acid-base extraction works, consider the extraction of a water-insoluble carboxylic acid (RCO2H) from a toluene solution containing a mixture of neutral organic compounds. The carboxylic acid, although virtually insoluble in water, can be extracted from the toluene (organic) solution into the aqueous phase by extracting with a solution of sodium bicarbonate. The basic aqueous sodium bicarbonate solution will react with the carboxylic acid to give a water-soluble carboxylate salt.
This salt will move into the aqueous solution, leaving the neutral organic compounds behind in the nonpolar toluene.
The carboxylic acid has been extracted from the organic layer, but now it is present in solution as the carboxylate salt. After the layers have been separated the carboxylic acid is regenerated using another acid-base reaction. Acidifying the basic solution with a mineral acid protonates the carboxylate ion, regenerating the carboxylic acid which has limited solubility in the aqueous solution. The isolated carboxylic acid product can then be recovered either by filtration or by extracting the carboxylic acid into fresh toluene and evaporating the solvent.
Carboxylic acids are strong enough acids (pKa 3-5) to be neutralized to water-soluble salts by reaction with a weakly basic sodium bicarbonate solution. The much less acidic phenols (pKa 8-10) will not react with sodium bicarbonate solution; phenols can be deprotonated to give water-soluble phenolate salts by extracting with an aqueous solution of a stronger base such as sodium hydroxide. The difference in the acidity of these two acidic functional groups allows them to be separated from each other in a mixture by using the proper alkaline solution in the acid-base extraction.
Organic amines, R3N, are bases that can be removed from an organic solution by extracting them with aqueous acidic solutions to form water-soluble ammonium salts. Once the ammonium salt has been
extracted into the aqueous phase and the organic and aqueous layers have been separated, the free amine can be regenerated from the ammonium ion by treating the aqueous solution with a base. This deprotonates the ammonium ion to give the less water-soluble free amine which can be collected either by filtration or by extracting the amine into fresh toluene and evaporating the solvent.
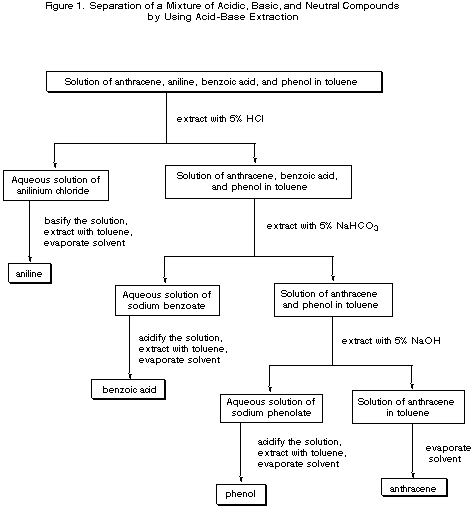
As an example of the utility of acid-base extractions in separating the components of a mixture, consider the separation of a mixture of anthracene, aniline, benzoic acid, and phenol.

The mixture is dissolved in toluene and placed in a separatory funnel, where it is first extracted with a 5-10% aqueous solution of HCl. This extraction removes aniline from the organic phase as the water-soluble anilinium chloride salt, leaving the other three components in the organic solution. The layers are separated and the organic solution is then extracted using a 5% aqueous solution of NaHCO3. This removes the benzoic acid from the organic solution as sodium benzoate but leaves the less acidic phenol behind in the organic layer. The layers are separated again, and now, by extracting the organic phase with 5% NaOH, phenol can be removed from the organic phase as the water-soluble sodium phenolate salt, leaving the neutral anthracene as the sole compound in the organic phase. Each component can now be isolated from the solution in which it is the sole component by using either a second extraction followed by solvent removal, or by neutralization and precipitation of the solid (See Figure 1).
When the experimental procedure calls for washing a solution, that means an extraction. An organic layer containing the desired material may be washed with aqueous sodium bicarbonate to remove acids, or washed with water to remove water-soluble impurities. An aqueous solution containing a desired organic salt may be washed with ether to remove unwanted neutral organic impurities. The liquid used for washing is normally the one that you don't intend to keep. A solvent used for extraction is normally the layer you save, containing the desired compound, but the operation is the same.
Using a separatory funnel
The choice of apparatus for an extraction is determined by the volumes of the solution being extracted and the extracting solutions. Typical extractions in the laboratory are done in a separatory funnel, while microscale extractions are done in a conical vial.
A separatory funnel is shown in the following figure. The funnel is fitted with a stopcock and a glass stopper. Make sure that the hole in the stopcock and the hole in the separatory funnel line up and that the stopcock fits snugly and turns smoothly without leaking. If the stopcock is made of glass, it should be lubricated with a small amount of stopcock grease so that it does not stick. Most newer stopcocks are made of Teflon® and need no grease. A small metal clip, rubber ring, or Teflon® nut with a washer and a ring at the end holds the stopcock in place. The stopper should also fit snugly. Avoid using stopcock grease on the stopper since the grease might dissolve in the organic solvent and contaminate the solution. For storage, remove the stopper and the stopcock, or wrap each with paper before inserting them into the funnel to prevent sticking.
A separatory funnel is top heavy, especially when nearly filled with liquid. Standing it upright in a beaker is a precarious situation at best. The separatory funnel should be supported in an iron ring of proper size attached to a ring stand. Before adding any liquid to theseparatory funnel, make sure that the stopcock is closed.
One of the most common errors in extraction is pouring a liquid into the separatory funnel without first making sure that the stopcock is closed. Oops! There goes your compound onto the benchtop.
Add the two immiscible liquids for the extraction through a funnel into the separatory funnel, filling the separatory funnel only about three-fourths full to allow room for mixing. Place the stopper in the separatory funnel and, holding the stopper firmly in place with the index finger of one hand, remove the separatory funnel from the iron ring.

Invert the funnel, pointing the stem up away from you (but not at someone else!), and carefully open the stopcock to vent any gases. You may hear a hissing sound as the gases are released through the stopcock. Close the stopcock and gently swirl the two liquids together to mix them. Vigorously shaking the contents is not only unnecessary but may prove deleterious by creating emulsions. It is common for pressure to build up in the separatory funnel during mixing, especially when using a volatile solvent such as ether or when neutralizing acids with carbonate or bicarbonate salts (that react with the acid to form CO2 gas) so it is necessary periodically to stop mixing and open the stopcock to vent any pressure that has built up. After mixing is complete, close the stopcock and place the separatory funnel in the iron ring, remove the stopper and allow the layers to settle and separate; a clean interface should form between the two layers.
Place an Erlenmeyer flask or a beaker under the funnel, open the stopcock and drain the lower layer into the beaker or flask. As the interface between the two solvents approaches the stopcock, slow the rate of draining by adjusting the stopcock; you want to remove all of the lower layer and retain all of the upper layer in the separatory funnel after draining.
If you are going to do another extraction on the top layer, leave it in the separatory funnel and add more of the next extracting solvent to the separatory funnel. If, however, you are going to do another extraction on the lower layer, pour the remaining upper layer out the top of the separatory funnel into a flask and then return the lower layer to the separatory funnel for the next extraction.
Which layer is which?
A common problem that you may face in doing extractions is trying to determine which one of the two layers in the separatory funnel is the aqueous layer and which one is the organic layer. The heavier layer (i.e., the more dense liquid) is the lower layer, of course, but some organic liquids (e.g., benzene, diethyl ether, ethyl acetate) are lighter than water and some (e.g., chloroform, dichloromethane) are heavier than water, so, depending on the solvents being used, the organic phase might be the upper or the lower layer in the separatory funnel. You could look up the densities of the two liquids to determine which is greater, but sometimes a high concentration of dissolved substances in the aqueous phase can cause it to be more dense than expected and to be found as the lower layer in the separatory funnel, even when extracting with an organic solvent with a density greater than water’s. If you are unsure about which layer is which, carry out a simple test: mix a small sample of each layer with a few drops of water in a test tube and see if the two liquids are miscible; the aqueous solution will dissolve the water drops but the organic solution will be immiscible with the added water.
The Golden Rule of Extraction is: Never throw a layer away until the end of the experiment, or until you are absolutely certain that you no longer need it. The most common mistake made during extraction is throwing the wrong layer down the drain or in the waste bottle, which means loss of material and starting the experiment over from the beginning.
Emulsions
The bugaboo of extractions is emulsions, foggy-looking mixtures containing tiny droplets of one liquid suspended in another that do not separate easily. If time permits, patience is the best solution; let the mixture stand until the emulsion breaks up and the two layers clearly separate. Sometimes adding salt or a saturated salt solution will help break up the emulsion, as will adding a little lighter or heavier solvent to the organic layer to increase the difference between the densities of the two layers. The best way to deal with emulsions is to avoid them. Gently swirling the contents of the separatory funnel during mixing instead of vigorously shaking them will help prevent emulsions from forming in the first place.