Abstract
The hitherto inaccessible nitroolefin (VII), which is the most convenient intermediate for the synthesis of the psychotropic amine (VIII), has been obtained by the direct condensation at high pressures (up to 1500 MPa) of piperonal (V) with 1-nitropropane (VI). The structure of (VII) was confirmed by direct synthesis from pyrocatechol (X). The amine (VIII) was obtained in three steps from (VII). This synthesis of (VIII) is shorter than that previously reported.
The condensation of aromatic aldehydes (I) with nitroalkanes (II) to give β-nitrostyrenes (III) (the Henry reaction) is an important step in the synthesis of β-phenylethylamines and their analogs (IV), which are biogenic amines. With some (I) and (II), however, the yields of (III) are too low, which makes the preparation of compounds (IV) by this method difficult1:
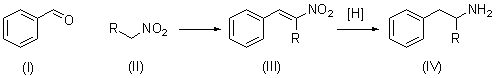
The principal aim of the present investigation was to examine the factors governing the condensation of piperonal (V) with 1-nitropropane (VI), which, according to literature reports, is too slow, gives large amounds of by-products, and is suitable for the preparation of large amounts of the required (VII)2. The development of a simple route for the preparation of (VII) from (V) and (VI) is important, since (VII) is the most convenient intermediate for the preparation of (VIII), a compound having unique psychopharmacological properties, and which is currently obtained by a rather laborious, multistage process2. For this reason, the direct synthesis of (VIII) from (VII) is of great practical interest:

Aldol-crotonic condensations are known to be accelerated considerably by pressure3. It would therefore be expected that it would also modify the rate of the reaction of (V) with (VI), so that the preparation of (VII) in preparative quantities might prove possible.
Table 1.
Dependence of the Yield of (VII) on
Conditions of Reaction of (V) with (VI)

Our studies of the condensation of (V) with (VI) have shown (Table 1) that pressure does indeed markedly accelerate this reaction and, in addition, improves its selectivity, under optimum conditions (Exp. 25, Table 1), the yield of (VII) reaching 65%. The effect of pressure on the yield of (VII) is well shown by a series of experiments (Table 1, Expts 1-15, and Fig. 1), carried out at the same temperature and time of reaction of (V) with (VI). The broken lines (a and b) in Fig. 1 indicate the phase transition pressures (due primarily to crystallization of the acetic acid) in the system (V) + (VI) + AcOH + AcONH4, as found from the decrease in volume of the mixture near these pressures4.
Fig 1.
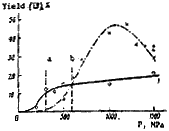
Plot of pressure
vs. VII yield in
the system AcOH-
NH4OAc-V-VI at
20°C1 & 50°C2.
The vertical lines
indicate phase
transitions at
20°Ca & 50°Cb.
As seen in Fig. 1, in the crystallization region the yield of (VII) increases, but on further increase in pressure the change is not as great. At 50°C, the yield curve shows a maximum at around 1200 MPa due, it appears, to further changes in the phase state of the system.
The solvent and catalyst used also have a considerable influence on the yield of (VII). The best catalysts were primary amines, and the best solvent acetic acid. The selectivity of the condensation of (V) with (VI) at high pressures usually increased, but it did not prove possible to eliminate entirely undesirable reactions, which always occurred, especially at elevated temperatures. One of the by-products, 3,4-methylenedioxybenzonitrile (IX), isolated by column chromatography and identical with that described, is probably formed, like other nitriles, from aromatic aldehydes and (VI)5.
The structure of (VII) was also confirmed by another, more laborious synthesis (X) → ... → (VII). The physicochemical properties of the (VII) obtained by the two methods were identical.

The availability of (VII) enabled us to develop a simple, three-step synthesis of (VIII) involving reduction of (VII) with LiAlH4, formulation of the amino-group and subsequent reduction with LiAlH4. Isolation of the compounds at the intermediate stages was unnecessary. The physicochemical properties of (VIII) were in agreement with those reported2. The availability of (VII) by direct condensation of (V) with (VI) therefore makes this method for the synthesis of (VIII) more convenient than that reported previously2.
Experimental
Melting points were determined on a Boetius hot plate. PMR spectra were obtained on Bruker WM-250 and Jeol 90 FQ spectrometers, in chloroform (internal standard HDMS). Chemical shifts are given on the δ scale relative to TMS (δ HDMS = 0.055 ppm). 13C NMR spectra were obtained on a Bruker AM-300 (in chloroform, δ from TMS), and mass spectra on a Varian MAT CH-6.
1,2-Methylenedioxybenzene (XI)
Obtained as in Ref 7, yield 73%, bp 173-175°C, cf. Ref 7.
3,4-Methylenedioxybromobenzene (XII)
Obtained as in Ref 8, yield 78%, bp 123-124°C (22 mm), cf. Ref 7.
1-(3,4-methylenedioxyphenyl)-1-butene (XIII)
To 9.0 g (0.36 mole) of magnesium turnings in 300 ml of dry THF was added a small crystal of iodine and 10.0 g (0.05 mol) of (XII). The mixture was heated with stirring to -40°C, and 30 min after the reaction commenced a further 62.3 g (0.31 mole) of (XII) was added, keeping the temperature below 50°C. The greenish solution of the Grignard reagent was cooled to -15°C, 25.9 g (0.36 mole) of freshly-distilled n-C3H7CHO added dropwise (below -10°C), the mixture boiled for 30 min, cooled, decomposed with aqueous ammonium chloride, and evaporated under reduced pressure. The residue was extracted with ether, dried over Na2SO4, the ether removed, and 100 mg of KHSO4 added to the residue. This mixture was heated slowly to 220°C under a vacuum of 10 torr in an oil bath. The distillate [a mixture of water and (XIII)] was dried over Na2SO4 and redistilled in vacuo, bp 135-137°C (10 mm), yield 34.8 g (55%), cf.2.
1-(3,4-Methylenedioxyphenyl)-2-nitro-1-butene (VII)
To a mixture of 8.8 g (0.05 mole) of (XIII), 4.8 g (0.061 mole) of dry pyridine, and 15 ml of acetone, cooled to 0°C, was added portionwise with stirring a solution of 9.8 g (0.05 mole) of tetranitromethane in 20 ml of acetone. The mixture was kept at 0°C for 1.5 h, poured into 100 ml of water, shaken, and ether added, followed by a solution of 3.44 g of 85% KOH (0.052 mole) in 60 ml of water. The ether layer was separated, the aqueous layer extracted with ether, washed with water, dilute sulfuric acid and water, and evaporated under reduced pressure. The residue was filtered, washed with cold methanol, and dried to give 8.29 g (75%) of bright yellow (VII), mp 66-66.5°C (from methanol) [spectral data dropped - Stalin]
High-pressure experiments:
A portion of a mixture of 0.068 mole of (V), 0.132 mole of (VI), and 0.056 mole of AcONH4 (or amine) and 0.744 mole of acid (see Table 1) was placed in a Teflon ampul of capacity 1-1.5 ml, and the conditions given in Table 1 established. When the reaction was complete, the mixture was poured into water, extracted with chloroform, washed with water, dried over Na2SO4, and evaporated. The residue was analyzed for (VII) by PMR. The (VII) was isolated by crystallization of the residue from methanol.
Isolation of 3,4-methylenedioxybenzonitrile (IX)
The worked-up reaction mixtures from the high-pressure experiments were applied to a short column of silica (100-250 µm), eluted with benzene until the yellow band (VII) began to be eluted, and the colorless eluate evaporated and the residue crystallized from methanol to give (IX), mp 90-93°C, M+ 147 m/z. [spectral data dropped - Stalin]
1-(3,4-Methylenedioxyphenyl)-2-methylaminobutane (VIII)
To a suspension of 17.5 g (0.46 mole) of LiAlH4 in 300 ml of dry THF was added dropwise with cooling to a solution of 23.74 g (0.107 mole) of (VII) in 90 ml of THF. The mixture was boiled for 1 h, cooled, decomposed with aqueous THF, stirred for 1 h, filtered, and the residue washed several times with ether and the filtrates evaporated. To the resulting oil (19.16 g) was added 100 ml (an excess) of ethyl formate, and the mixture boiled for 7 h, evaporated, and residue dissolved in ether, washed with cold 5% HCl and water, dried over MgSO4, and evaporated. The residue (an orange oil) was dissolved in 100 ml of THF, and the solution added dropwise with stirring to a suspension of 14 g (0.37 mole) of LiAlH4 in 400 ml of THF. The mixture was boiled for 1 h, cooled, decomposed with aqueous THF, filtered, the solid washed with THF, the filtrates evaporated, and the residue redissolved in 180 ml of absolute ethanol. Concentrated hydrochloric acid (6 ml) was then added, diluted with ether to a volume of 1400 ml, and the crystalline solid filtered off to give 7.93 g [30% on (VII)] of (VIII) hydrochloride, mp 146.5-149°C (propan-2-ol). [spectral data dropped - Stalin]