Abstract
Synthesis of 2-amino-1-(4-methoxyphenyl)-propane (II) starting from p-anisaldehyde (1) in 67% overall yield is described. Key reactions involved Horner–Wadsworth–Emmons olefination of 1 to form the ester 2 and Hoffmann degradation of amide 5 to obtain the amine II.
Introduction

β-Arylethylamine functionality is essential to many amphetamine drugs that offer high selectivity for 2-adrenoceptors1. A variety of 2-adrenoceptor agonists have been prepared for treatment of asthma and chronic bronchitis2a-d. Among them formoterol (I) offers high selectivity for 2-adrenoceptors and has excellent safety, tolerance profile and has been commercialized in racemic form3a-d. Synthesis of I requires preparation of 2-amino-1-(4-methoxyphenyl)-propane (II) by a process which is amenable to scale up, avoids use of hazardous reagents, elevated temperature and pressure. Synthetic procedures to prepare β-phenyl ethylamine II describes Henry reaction of p-anisaldehyde (1) to obtain the key intermediate β-nitrostyrene4 III followed by hydrogenation at high pressure in presence of Pd/C5 or Red-Al4b or by refluxing with LiAlH4 in Et2O4f to obtain the amine II in 70–85% yield in one approach and in the second III was converted to the keto compound IV by reaction with Fe/aq. HCl4a, or NaH2PO2·H2O/Raney–Ni at pH 56 or HClO47 or Zn–TFA8. The keto compound IV was converted to the corresponding oxime V9b by reaction with NH2OH·HCl followed by reduction with Raney–Ni at high pressure4a to obtain the amine II in 40–43% overall yield.
We report herein a simple laboratory method for the preparation of amine II.
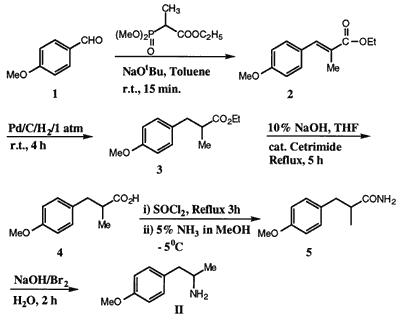
Horner–Wadsworth–Emmons reactionThe Horner–Wadsworth–Emmons reaction of 1 with ethyl (2-dimethoxyphosphinyl)-2- propanoate10 (1.1 mol equiv.) and NaOtBu (1.5 mol equiv.) in toluene at room temperature for 15 min gave the α,β-unsaturated ester 2 which on catalytic hydrogenation (1 atm.) with 10% Pd/C in MeOH for 4 h gave the saturated ester 3, while progress of the reaction was monitored by HPLC (experimental). Hydrolysis of 3 with aq. NaOH (20%) and a catalytic amount of cetrimide at reflux temperature for 5 h gave the acid derivative 4. Compound 4 was transformed to the amide 5 (mp: 119–121°C) by reaction with SOCl2 followed by quenching with methanolic ammonia solution at -5°C. Hoffman degradation of amide 5 by reaction with NaOBr at 70°C for 2 h gave the amine II in 67% overall yield from p-anisaldehyde (1).
Experimental
Ethyl-3-(4-methoxyphenyl)-2-methyl-2-propenoate (2)
To a solution of p-anisaldehyde 1 (32 g, 0.24 mol), toluene (160 mL) and ethyl-(2-dimethoxyphosphinyl)-2-propanoate (58.8 g, 0.28 mol) was added NaOtBu (33.6 g, 0.35 mol) at 0°C under N2 atmosphere during half an hour. The reaction mixture was brought to room temperature and stirred for 15 min. Progress of the reaction was monitored by TLC [AcOEt:Hexane; 1:4] from the appearance of a faster moving spot. After completion of the reaction, the reaction mixture was neutralised with 10% aq. HCl. The toluene layer was separated, washed with water (2×100 mL), dried (Na2SO4) and concentrated to obtain the title compound 2 as a syrup in quantitative yield (51.6 g).
Ethyl-3-(4-methoxyphenyl)-2-methyl-propanoate (3)
To a solution of 2 (32 g, 0.145 mol) in MeOH (160 mL) was added Pd/C 10% (1.0 g) and subjected to hydrogenation (1 atm) for 4 h. Progress of the reaction was monitored by HPLC (Eluant: 70% CH3CN in H2O, 2 had a retention time of 8.3 min and compound 3 of 7.3 min). After completion of the reaction, the catalyst was filtered off and the filtrate was concentrated to obtain the title compound 3 as a syrup in quantitative yield (31.8 g).
3-(4-Methoxyphenyl)-propanoic Acid (4)
To a solution of 3 (25 g, 0.11 mmol) in THF (93 mL) was added NaOH (8.96 g in 46 mL H2O) (0.22 mol), Cetrimide (0.2g) and refluxed for 5 h. The progress of the reaction was monitored by TLC [AcOEt:Hexane; 2:3] from the appearance of a slower moving spot. After completion of the reaction, THF was removed on a rotary evaporator and the reaction mixture was acidified with 10% aq. HCl and extracted into CH2Cl2 (125 mL). The organic layer was separated, washed with water (2×100 mL), dried (Na2SO4) and concentrated to obtain the title compound 4 as a syrup in quantitative yield (21.5 g).
3-(4-Methoxyphenyl)-2-methyl-propanamide (5)
A solution of 4 (20 g, 0.10 mol) in SOCl2 (14.6 mL, 0.20 mol) was heated to reflux for 2 h. After completion of the reaction, excess thionyl chloride was distilled off to obtain a residue which was dissolved in MeOH (60 mL), cooled to -5°C under nitrogen atmosphere and a 5% solution of ammonia in MeOH (3.50 g in 70 mL MeOH, 0.20 mol) was added dropwise under efficient stirring at -5°C. After completion of the reaction, methanol was removed on a rotary evaporator to obtain a solid which was filtered, washed with water (100 mL) and dried to obtain the title compound 5 in quantitative yield (19.30 g); mp 119–121°C.
2-Amino-1-(4-methoxyphenyl)-propane (II)
To a stirred solution of sodium hypobromite [Br2 (4.6 mL, 0.09 mol), NaOH (18.72 g, 0.48 mol) in 150 mL water] was added a finely divided amide 5 (15 g, 0.078 mol) at 0°C during 10 min. The reaction mixture was heated to 70°C for 2 h. The progress of the reaction was monitored by TLC (BuOH:EtOH:NH3, 7:2.6:0.3 mL). After completion of the reaction, the reaction mixture was extracted into CH2Cl2 (75 mL). Organic layer was separated, washed with water (2×75 mL), dried (Na2SO4) and concentrated to obtain the title compound II as a syrup (9.6 g, 75%) that exhibited comparable 1H-NMR spectrum with that reported in literature4f,11a. For the purpose of characterisation II was also converted to the corresponding hydrochloride11b [10.5g, 65%, mp 208–210°C (lit.4a 208°C)] by treating with 5% HCl in MeOH.