I wanted to synthesize some aspirine the other day, using the following igredients:
- 11.5 g 3,4,5-trimethoxybenzaldehyde (M = 196.2 g/mol, purity > 98%)
- 5 mL nitroethane (M = 75.0 g/mol, d = 1.05 kg, purity > 97%)
- 30 mL EtOH (absolute)
- 168 mg NaF (M = 42 g/mol, purity > 99%)
I brought some of the 3,4,5-trimethoxybenzaldehyde in a RB flask (250 mL) and added 15 mL EtOH. I then added 5 mL nitroethane, followed by another 15 mL EtOH. I dropped the magnetic stirbar in the RB and while stirring, the NaF was added. After half an hour of stirring, it looked clear to me that the 3,4,5-trimethoxybenzaldehyde dissolved very slowly, so I heated the RB a little bit (a little bit, i.e. not refluxing). After another 2 hours, I stopped the stirring. The 3,4,5-trimethoxybenzaldehyde was dissolved completely, the reaction mixture was pale yellow in colour. I then decanted the reaction mixture in 100 mL cool demineralized water. Pricipitation commenced (PREP1). I decanted the water in another 50 mL cool demineralized water where precipitation continued (PREP2).
PREP1 was hard and yellowish white in colour. PREP2 was bright white. I dissolved some crystals in iso-octane and injected a 1 µL amount on GC/MS. The resuling chromatogram and mass spectrum are shown in the following figure.
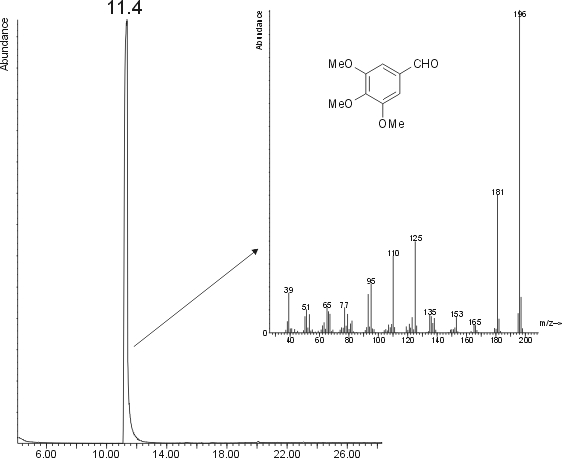
As you see, the main peak (there is only one, you cannot miss it) is a compound with M
+ = 196 amu. The mass spectrum gives a 1% match for aspirine, but a 98% for 3,4,5-trimethoxybenzaldehyde

.
I don't know how to give these data a good interpretation... Or it is my failure (has been several months since I performed an organic synthesis), or NaF cannot be used. I'd appreciate it if another bee tries it out as well.
geh in die knie. wackle mit den hueften. klatsch in die haende. und tanz den mussolini.

