Synthesis and pharmacology of potent 5-HT2A receptor agonists which have a partial N-2-methoxybenzyl structureRalf HeimPhD thesis, FU Berlin, Germany
(
http://www.diss.fu-berlin.de/2004/81/indexe.html)
Abstract: Serotonin-2A(5-HT
2A) receptors play a significant role in causing hallucinations and also in the development of psychological disorders such as schizophrenia and depression. The 5-HT
2A receptor subtype is also involved in the mediation of cognition and emotion. Subtype selective compounds which show (partial)agonistic effects at 5-HT
2A receptors are used in basic medicinal and pharmaceutical research as important experimental tools for the investigation of complex physiological and pathophysiological processes involving the neurotransmitter serotonin (5-HT). A new structure-activity concept for 5-HT
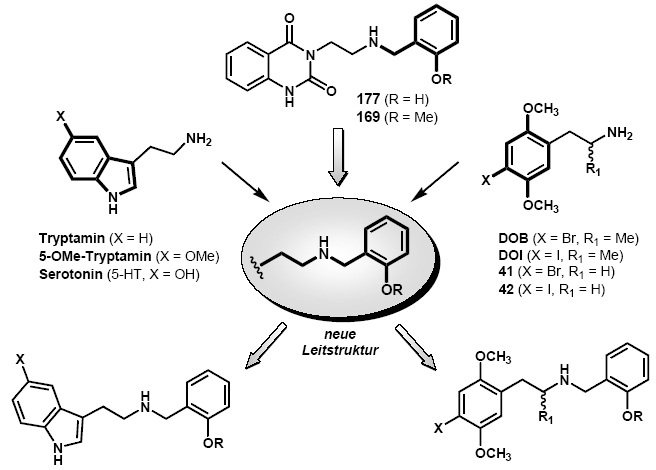
2A receptor agonists was developed, following the discovery of unexpected partial agonistic activity of a molecular fragment of the classical 5-HT
2A receptor antagonist ketanserin, belonging to the 3-(2-aminoethyl)-2,4(1H,3H)quinazolindione class. This new structure-activity concept results in a dramatic increase of the agonistic activity of 5-HT
2A receptor agonists with primary amine functionality by insertion of a N-2-methoxy- or N-2 hydroxybenzyl functional group and could also be applied to tryptamines and phenylethylamines. The key step during the synthesis of the N-substituted 3-(2-aminoethyl)-2,4(1H,3H)quinazolindione-type amines is nucleophilic ring opening of the tricyclic 2,3-dihydro-5-oxo-5H-oxazolo-[2,3-b]-quinazoline (
95) with various primary amines. The most potent compound in this series is the 2-methoxybenzyl derivative
169 (pEC
50 = 6.58, Emax = 49 %) which displays higher than 250 fold increase of the partial agonist activity compared to the lead structure
93 with a primary amine functional group. The synthesis of the N-benzylated 2-(1H-indol-3-yl)ethylamine derivatives proceeded via indirect reductive alkylation of either tryptamine or 5-methoxytryptamine. The synthesis of the N-2-methoxybenzyl substituted DOB- and DOI-type phenylethylamines occurred via the following sequence: Henry-reaction, followed by reduction of the nitrostyrenes and finally reductive alkylation of the primary amines. In an alternative synthetic pathway a Cbz protected aminoethyl sidechain was inserted in only one step via a hydroboration/Suzuki coupling reaction. The most potent compounds in the phenylethylamine series are the 4-bromo and the 4-iodo derivatives
230,
231 and
235,
236 respectively, which display sub-nanomolar activity in an in vitro vessel assay. The pEC
50 values of these partial agonists are in the range of 9.58 (0.26 nmol × L
-1) up to 10.13 (74 pmol × L
-1), displaying potencies up to 1350 times higher than that of the reference agonist serotonin, and almost 106 times higher in comparison to the starting lead structure
93. Furthermore the synthesis of the highly potent tetrahydrobenzodifuranes
270 and
271 (pEC
50 = 9.87 resp. 10.15), rigid analogues of the 2,5-dimethoxyphenylethylamines
230 and
231, provide invaluable insight into the active binding conformation of these drugs. To investigate the stereoselectivity of the 5-HT
2A receptor binding site, both enantiomers of the chiral quinazolindiones 298 and the chiral phenylethylamines
304 and
305 were synthesised in very high optical purity (ee > 99 %). The chiral key building block 1-(2-methoxyphenyl)ethylamine (
288) was synthesised via highly diastereoselective reductive amination (d.r. = 97 : 3) to give both enantiomers of
288 in excellent enantiomeric purity (ee > 99 %). An enzymatically catalysed kinetic resolution of (±)-N-[1-(2-methoxyphenyl)ethyl]oxalic acid octylester (
291) with immobilised lipase B of Candida antarctica is also described.