Anyone with access to reference #14 and #15?Synthesis and analgesic activity of novel N-acylarylhydrazones and isosters, derived from natural safroleEur. J. Med. Chem. 35, 187-203 (2000)ChemistryThe safrole derived target compounds were prepared from Piperonal
15, obtained in ca. 75% overall yield from the natural safrole
7, using base catalysed isomerization of the double bond
[14] followed by oxidative cleavage
[15]. Employing the oxidative Yamada’s procedure
[16] compound
15 was ‘one-pot’ converted, in 90% yield, to the corresponding methyl ester
16 by treatment with 2.6 eq. of KOH and 1.3 eq. of iodine in methanol at 0 °C.
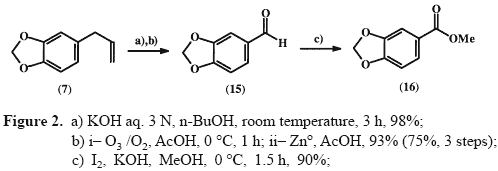 Methyl 3,4-methylenedioxybenzoate 16
Methyl 3,4-methylenedioxybenzoate 16To a solution of piperonal
15 (0.45 g, 3.0 mmol) in absolute methanol (4 mL) cooled at 0°C, were successively added methanolic solutions (each 3 mL) of iodine (1.00 g, 3.9 mmol) and KOH (0.44 g, 7.8 mmol) at 0°C. After stirring for 1.5 h at 0°C, small amounts of saturated NaHSO3 solution were added until the disappearance of the brown colour. Next, the methanol was almost totally evaporated under reduced pressure. To the residue was added water, and the desired methyl 3,4-methylenedioxybenzoate 16 was obtained by filtration, in 90% yield, as a white solid, mp 53°C.
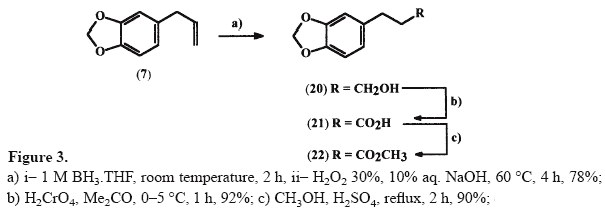 3-(3',4'-Methylenedioxyphenyl)propan-1-ol 20 [15]
3-(3',4'-Methylenedioxyphenyl)propan-1-ol 20 [15]A solution of 0.92 g (5.7 mmol) of safrole
7 in 25 mL of dry THF, at room temperature, was treated with 6 mL (6 mmol) of a 1 M solution of BH3.THF, under nitrogen atmosphere. The resulting solution was stirred at room temperature for 2 h. Then, methanol was added dropwise until no further gas was evolved, and 2.4 mL of a 10% aqueous NaOH solution (6 mmol) and 2.4 mL of 30% aqueous H2O2 solution were added at 0°C. The suspension formed was maintained at 60°C for 4 h. After cooling, the reaction mixture was partitioned between ethyl ether (20 mL) and water (10 mL), followed by separation of the organic phase. The aqueous layer was further extracted with ethyl ether (3x10 mL) and the organic extracts were combined, treated with 10 mL of a 10% aqueous HCl solution and submitted to the usual work-up which furnished compound
20 as a yellow oil, 78% yield (R
f ethyl acetate: hexane 30% = 0.35).
3-(3',4'-Methylenedioxyphenyl) propanoic acid 21 [15]An 8 N solution of Jones reagent
[19] (ca. 10 mL) was added dropwise to a solution of 1.15 g (6.4 mmol) of alcohol
20 in 10 mL of acetone at 0°C. After 1 h 10 mL of isopropanol was added and the reaction mixture was filtered. The filtrate was concentrated under reduced pressure to obtain a residue which was diluted with 50 mL of a mixture of diethylether/water (2:1). The organic layer was separated, dried and evaporated to give 1.14 g (92%) of the acid
21 as a white solid, mp 81–82°C (R
f acetone:toluene 30% = 0.47).
Methyl 3-(3',4'-methylenedioxyphenyl) propanoate 22To a solution of 0.580 g (3 mmol) of the acid
21 in 10 mL of MeOH were added 0.3 mL of concentrated H2SO4 and the reaction mixture was refluxed for 2 h, when analysis by TLC indicated the end of the reaction. Then, the solvent was concentrated at reduced pressure and the residue obtained was neutralized with 10% aqueous NaHCO3 solution. Extraction of the aqueous phase with ethyl acetate (3x10 mL), followed by the usual work-up of organic layers, furnished 0.62 g (90%) of the corresponding ester derivative
22 as a light yellow oil, (R
f acetone:toluene 30% = 0.81).
References:[14] Barreiro E.J., Lima M.E.F., J. Pharm. Sci. 81 (1992) 1219–1222.
[15] Barreiro E.J., Costa P.R.R., Coelho F.A.S., de Farias F.M.C., J. Chem. Res. (M) (1985) 2301–2332.
[16] Yamada S., Tetrahedron Lett. 33 (1992) 4329–4332. DOI:
10.1016/S0040-4039(00)74252-3
[19] Hudlicky M., Oxidations in Organic Chemistry, American Chemical Society, Washington DC, 1990, p. 273.

