This procedure is very suited to our needs, it was more tuned up than the former
http://www.orgsyn.org/orgsyn/prep.asp?prep=cv6p0890
poix posted all above this thread. Both procedure use the same silver/persulfate to generate Ag2+
in situ which oxyde a suitable alkyl chain to form a radical that will get quenched by the quinone. There are two notable differences here against the former:
-first: the alkyl chain is not an acid which loss CO2 to generate the alkyl radical minus one C based on the acid (ex: butyric -> CH3CH2CH2. ) , but an ester of oxalic acid, which come from an alcohol with the same number of carbon than the alkyl chain to bee put in place. The oxalate is oxydatively hydrolised and generate a R. radical, where the former OH was, which will react with the quinone.
-second: here they solve the problem of poly alkylation of the quinone by various radicals by using a two phases mixture. This can bee used in the old orgsyn.org ref too, if you want to start with the acid in place of the oxalate. Higher yield and purity are achieved through such reaction medium change.
The true problem now is synthetising the allyl oxalate, as I said in another thread, it can bee done by transesterification of oxalate diethyl ester, then basic hydrolyse of one of the allyl ester to the free acid. Sadly the patents are in Japanese. Refs to make this allyl oxalate are
highly wanted. Well, at worst using some proper acid (like N-acetyl
beta-alanine or the acetal of acetoacetic acid) and this biphasic medium in place of the one phase of orgsyn ref will give rise to better yields and next to zero polyalkylation of the quinones. The convenience of this route gained a few points here, bees.
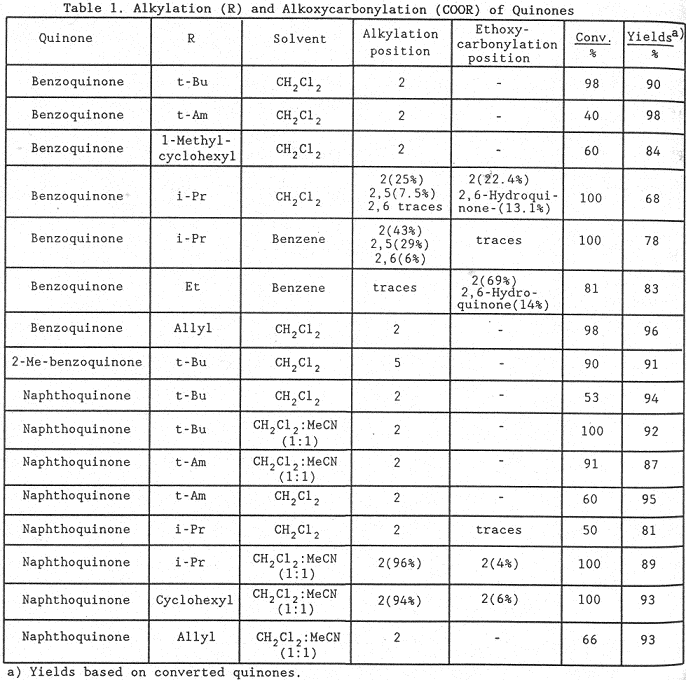
 Check the table: 96% for the allylation of quinone! Yeesh!
Check the table: 96% for the allylation of quinone! Yeesh! 
Here is the gem:
A New Selective Method for the Homolytic Alkylation and Carboxylation of Quinones by Monoesters of Oxalic Acid Fausta COPPA, Francesca FONTANA, Edoardo LAZZARINI, and Francesco MINISCI Chem. lett. 1992 7 1299Abstract:Alkyl and alkoxycarbonyl radicals were generated by oxidative decarboxylation of oxalic acid monoesters by persulfate; they were then utilized for the selective substitution of quinones.The oxidative decarboxylation of oxalic acid monoesters proved to be a very effective source of alkoxycarbonyl and alkyl radicals, useful for selective syntheses. The alkylation of heteroaromatic bases was described in the preceding Letter [1] and in a recent report [2] of a more expensive and less effective procedure.
Now we report how this radical source can be successfully utilized for the selective alkylation (Eq.1) or carboxylation (Eq.2) of quinones in a two-phase system. The results are shown in Table 1. With esters of tertiary or secondary alcohols, alkylation (Eq.1) mainly occurs, whereas with primary alcohols carboxylation (Eq.2) becomes the main process. With esters of allylic alcohols only allylation occurs and we expect a similar behaviour with esters of benzylic alcohols.
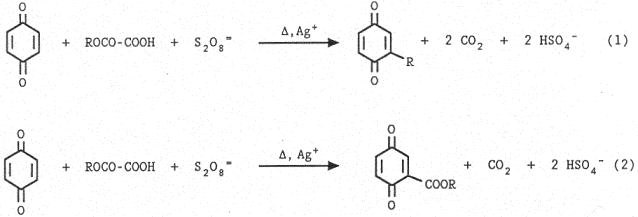
Operating in a two-phase system, constituted by water and an organic solvent, such as CH2Cl2 or benzene, is particularly important for minimizing polysubstitution, because the reaction products are generally more lipophilic than the starting quinones and are therefore preferentially extracted by the organic solvent, whereas the substitution reaction takes place in the aqueous phase. With quinones of very low solubility in water, such as the naphthoquinone derivatives, using two-phase system constituted by three solvents (CH2Cl2, CH3CN, and H20) improves the effectiveness of the reaction.
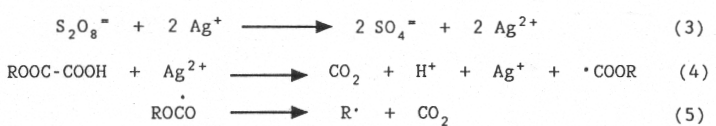
The mechanism of the reaction involves the following steps:
i) generation of the carbon-centered radicals (Eqs.3-5)
ii) addition to the quinone ring (Eq.6)
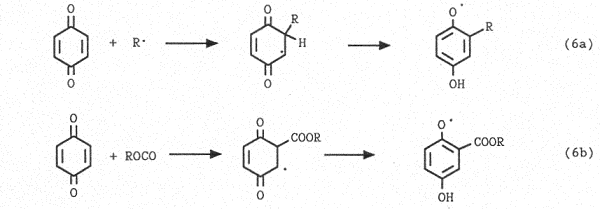
iii) oxidation of the radical adduct in a redox chain (Eq.7)
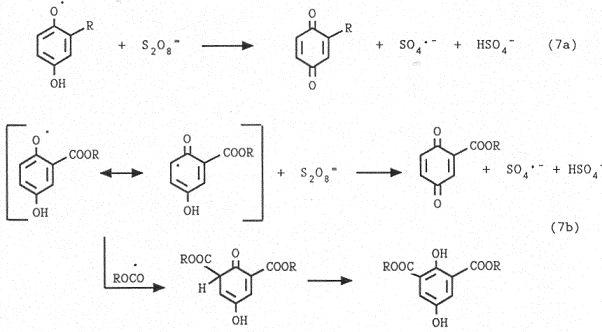
It is noteworthy that when alkoxycarbonylation (Eq.2) is the prevailing reaction, as in the case of the reaction between benzoquinone and ethyl monoester, a minor amount of 2,6-diethoxycarbonylhydroquinone is formed. We explain this result by the fact that the introduction of an alkoxycarbonyl radical, instead of an alkyl radical, on the quinone ring increases the redox potential of the resulting phenoxy radical and makes its oxidation by persulfate slower (Eq.7). This allows to reach stationary concentration of the phenoxy radical, suitable for acting as scavenger towards another alkoxycarbonyl radical (Eq.7b).
Considering that the reaction takes place in the aqueous phase, in which the solubility of the quinone is generally very low, that the ethyl radical is not formed in siqnificant amount and that the rate is given by the expression r = k [-COOR] [quinone], it follows that the rate constant for the addition of the ethoxycarbonyl radical to the quinone ring must be high (>10^6 M-1 s-1). The lower solubility of naphthoquinone explains its lower degree of alkoxycarbonylation compared to benzoquinone under identical reaction conditions.
A general experimental procedure is given:A solution of 10 mmol of monoester of oxalic acid and 5 mmol of quinone in 20 ml of the solvents reported in the Table was added to 20 ml of an aqueous solution containing 10 mmol of Na2S208 and 0.5 mmol of AgN03. The mixture was refluxed for 2 h, then the organic layer was separated, dried and analyzed by GC and GC/MS. The reaction products were isolated by flash-chromatography on silica gel and identified by comparison with authentic samples.[3] This is the first example, to the best of our knowledge, where the homolytic carboxylation, of quinones is achieved. On the other hand, the above described alkylation represents the only procedure so far known for the radical alkylation of quinones by alcohols, whereas alkylation by carboxylic acids has been reported by several groups.[4]
References:[1] F. Coppa, F. Fontana, E. Lazzarini, F. Minisci, and L. Zhao,
Chem.Lett., preceding paper.[2] H. Togo, M. Aoki, and M. Yokoyama,
Chem.Lett., 1991, 1691.[3] F. Coppa, F. Fontana, F. Minisci, M. C. Nogueira Barbosa, and E. Vismara,
Tetrahedron, 47, 7343 (1991) and references therein.
[4] Ref.3; D. H. R. Barton, D. Bridan, and S. Z. Zard,
Tetrahedron, 43, 5307 (1987); B. Lin, L. Gu, and J. Zhang,
Rec.Trav.Chim.Pays-Bas, 110, 104 (1991) and references therein.

