An Improved Preparation of 1-Methyl-4-Cyano-4-PhenylpiperidineD. Gnecco, J. L. Teran, R. G. Enríquez, W. F. Reynolds, C. MarazanoOrg. Prep. Proced. Int. 28(4), 478-480 (1996)Several synthetic approaches are available for the preparation of 1-methyl-4-cyano-4-phenylpiperidine (
1). These methods all involve refluxing the starting material in toluene or benzene for several hours which results in the formation of polymeric residues, making the purification process difficult and leading to low overall yields
1. Since
1 is a key intermediate for the synthesis of derivatives which exhibit potent narcotic-analgesic or antihypertensive effects
2, it would be highly desirable to have a better method for the synthesis of
1. The present work describes modifications that significantly improve its preparation.
Reaction of phenylacetonitrile and
bis-(
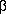
-chloroethyl)methylamine hydrochloride in anhydrous ether with sodium amide at -5°C afforded
1 in yields which were on average 30-40% higher than those previously reported
1.
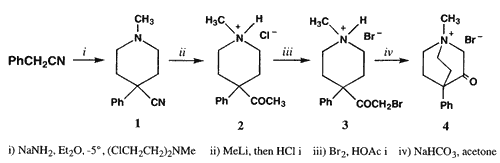
1-Methyl-3-keto-4-phenylquinuclidinium bromide (
4) and intermediates
2 and
3 were prepared from
1 according to Perrine's method
3.
Experimental Caution: Care should be exercised when preparing compound
1 due to the toxicity of
bis-(
-chloroethyl)methylamine hydrochloride and derivatives.
1-Methyl-4-cyano-4-phenylpiperidine (1) To
bis-(
-chloroethyl)methylamine hydrochloride (4.0g, 20.8 mmol) and sodium amide (3.3 g, 83.2 mmol) in a 250 mL round bottom flask in 50 mL of anhydrous ether with magnetic stirring and an inert atmosphere of Argon at -5°C, was added dropwise a solution of phenylacetonitrile (5 mL, 43.4 mmol) in 50 mL of anhydrous ether. After 50 min at room temperature, the reaction was complete. The mixture was cooled to 0°C and upon addition of 15 mL of H
2O, two phases separated. The organic phase was washed with a saturated solution of ammonium chloride until neutral. After drying over anhydrous sodium sulfate and removal of solvent in vacuo, a yellowish oil (7.90 g, 95%) consisting of a mixture of the free base of
1 with unreacted phenylacetonitrile was obtained. The crude product was dissolved in 10 mL of ethanol and 2 mL of concentrated HCl were added which afforded the hydrochloride of
1 (4 g, 80% based on reacted phenylacetonitrile) as colorless needles, mp 223-225°C. The hydrochloride of
1 (1 g) could be converted quantitatively into the free base (yellow oil).
Compound 1·HCl 1H NMR (DMSO-d6): 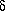
2.49 (m, 4H), 3.31 (s, 3H), 3.30 (br, t, 2H), 3.60 (d, J
gem=12.3 Hz, 2H), 7.60 (m, 5H).
References1. a) H. R. Le Sueur and P. Haas. J. Chem. Soc., 97, 967 (1910);
b) O. Eisleb and O. Schaumann, Deut. Med. Worrsch., 65, 967, Chem. Abstr., 36, 54653 (1939);
c) O. Eisleb, Ber., 74, 1443 (1941);
d) A. T. Nielsen, J. Org Chem., 31, 1053 (1966);
e) J. Carrol, A. N. Fergusson and J. B. Lewis, J. Org Chem., 31, 2957 (1966);
f) D. S. Watt, Tetrahedron, 24, 175 (1968).
2. a) E. Hong,
Patent US3860717
; Chem. Abstr., 82, 149531z (1975);
b) L. E. Mather and P. J. Meffin., Clin. Pharmacokinet., 3, 352 (1978); Chem. Abstr., 90, 40w (1979).
3. T. D. Perrine, J. Org. Chem., 22, 1484 (1957).

