Enantioefficient Synthesis of alpha-Ergocryptine: First Direct Synthesis of (+)-Lysergic Acid István Moldvai, Eszter Temesvári-Major, Mária Incze, Éva Szentirmay, Eszter Gács-Baitz, and Csaba SzántayJ. Org. Chem., ASAP Article, Web Release Date: August 3, 2004DOI:
10.1021/jo049209
Chemical Research Center, Institute of Biomolecular Chemistry, Hungarian Academy of Sciences, POB 17, H-1525 Budapest, Hungary, and Research Group for Alkaloid Chemistry of the Hungarian Academy of Sciences, Budapest University of Technology and Economics, Gellért tér 4, H-1521 Budapest, HungaryReceived May 11, 2004 Abstract:The first direct synthesis of (+)-lysergic acid (
2a) suitable for scale-up has been achieved by the following reaction sequence. Bromoketones
4d or
4g were allowed to react with amine
5 followed by deprotection, and the resulting diketone
6c was transformed into the unsaturated ketone (±)-
7 by the LiBr/Et
3N system. Resolution afforded (+)-
7, which was further transformed by Schöllkopf's method into the mixture of esters
2e and
2f. Upon hydrolysis the latter mixture afforded (+)-
2a. The peptide part of
alpha-ergocryptine (
1) was prepared according to the Sandoz method; the stereoefficiency, however, has been significantly improved by applying a new resolution method and recycling the undesired enantiomer. Coupling the peptide part with lysergic acid afforded
1. Having synthetic (+)-
7 in hand, we can claim the total synthesis of all the alkaloids which were prepared earlier from (+)-
7that had been obtained through degradation of natural lysergic acid.
Introduction"
Ergot alkaloids, of which lysergic acid is representative, are particularly important as they possess the widest spectrum of biological activity found in any family of natural products".
1 One of the biologically most important ergot alkaloids is -ergocryptine (1). Its semisynthetic derivative, the so-called bromocryptine, is one of the most widely used drugs in this family (e.g., as a prolactin inhibitor, or an anti-Parkinsonian).2 Great efforts have been devoted to the synthesis of ergot alkaloids during the second half of the last century. Conceptually, retrosynthetic cleavage of the central amide bond devides the problem into two parts, the synthesis of the lysergic acid and of the peptide dilactam moiety.
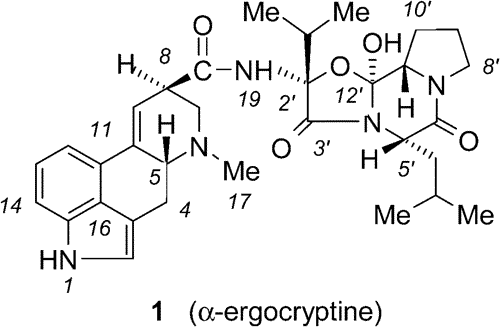 Scheme 1
Scheme 1The first synthesis of racemic lysergic acid was effected by Woodward and Kornfeld in 1956.
3,16 One of their main problems was to prepare ring C from the otherwise easily accessible 3-indolepropionic acid (
3), since the ring closure of the corresponding acid chloride occurred at the more reactive pyrrole ring instead of the benzene ring. Thus the Woodward group reduced the pyrrole ring, the amine was protected by benzoylation, and thereafter the ring closure took place regioselevtively as desired. The drawback of this approach is that sooner or later the pyrroline moiety must be reoxidized to a pyrrole ring. It is difficult to perform an enantioefficient synthesis as well, since the method involves introduction of an unnecessary chiral center by the reduction. The earlier described resolution of racemic compound needs further and rather inconvenient steps.
4 So far the total synthesis of (±)-
2a has been achieved by nine groups, but the number of publications dealing with the synthetic efforts is much higher. Among these approaches one can find about a dozen methods trying to construct the ergoline ring, some of which were successful; others remained at the level of attempt.
5 Seven of the nine successful syntheses used the reduced indoline derivative as the starting compound. Oppolzer et al.
3 performed the first total synthesis avoiding the reduction step, but their procedure again cannot be scaled up. A second approach to the racemic acid was published recently.
6We decided to construct the ergoline skeleton starting from indole, thus avoiding the reoxidation problem, and at the same time making an enantioefficient synthesis possible.
7An ideal starting material was the so-called Uhle's ketone (
4a) having the intact indole ring, although the original synthesis of
4a is rather tedious. Uhle commenced with acetylation and subsequent bromination, and he claimed that the derived bromo-derivative
4b could be subjected successfully to a substitution reaction with various amines.
9 Early in the seventies Bowman and co-workers reinvestigated a few results of Uhle's synthesis and established that one of the key steps, alkylation of several types of amines with
4b, always failed.
10 The approach starting from
4a by Stoll used the Stobbe condensation as a key step, but the reaction sequence could not be carried out,
11 thus the synthesis of lysergic acid starting from Uhle's ketone remained a challenge.
We decided to construct the ergoline skeleton starting from indole, thus avoiding the reoxidation problem, and at the same time making an enantioefficient synthesis possible.
7An ideal starting material was the so-called Uhle's ketone (
4a) having the intact indole ring, although the original synthesis of
4a is rather tedious. Uhle commenced with acetylation and subsequent bromination, and he claimed that the derived bromo-derivative
4b could be subjected successfully to a substitution reaction with various amines.
9 Early in the seventies Bowman and co-workers reinvestigated a few results of Uhle's synthesis and established that one of the key steps, alkylation of several types of amines with
4b, always failed.
10 The approach starting from
4a by Stoll used the Stobbe condensation as a key step, but the reaction sequence could not be carried out,
11 thus the synthesis of lysergic acid starting from Uhle's ketone remained a challenge.
Results and Discussion(1) Synthesis of (+)-Lysergic Acid. In 1994 the
N-pivaloyl derivative of Uhle's ketone (
4c) became easily accessible from 3-indolepropionic acid by Goto's method.
12 In connection with our attempt to find a reasonable total synthesis of ergoline skeleton we wished to reinvestigate the cyclization of ring D. We had reported the first successful reaction sequence to this end by applying an unprecedented intramolecular Stobbe condensation taking advantage of a lithium complex formed as an intermediate.
13 As a second approach, ring D of the tetracyclic skeleton was formed by an intramolecular Dieckmann condensation of a diester, obtained in a modified Reformatsky reaction of a properly substituted derivative of
4c, followed by elimination of water.
14 Neither of these methods, however, could be further elaborated to achieve (+)-lysergic acid.
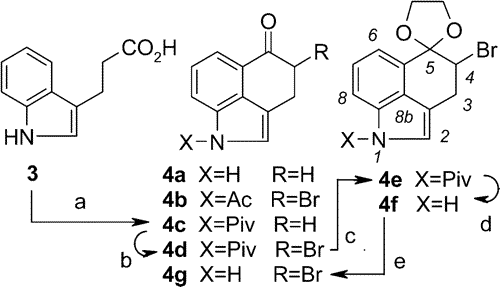 Scheme 2.
Scheme 2. Synthesis of 4-Bromo-Uhle's Ketone (
4g) from
3a a
Reagents and conditions: (a) (1) powdered KOH + Piv-Cl, CH2Cl2 + THF, (2) SOCl2, (3) AlCl3 + ClCH2COCl, CH2Cl2 (43%, overall);
(b) ref 15 (85%);
(c) HO(CH2)2OH, p-TSA, benzene, reflux, 6 h (81%);
(d) MeNH
2, CHCl
3, 10-15 °C, 3-4 h (88%);
(e) aq HCl (1 M), acetone, rt, 3 h (97%).
To our pleasant surprise we found that bromoketone
4d, contrary to the literature,
10 can be subjected to a substitution reaction with amine
5 providing us with the so far unknown, but much sought-after, even mistakenly claimed
9 product (
6a), if one has the patience to allow the reaction to proceed at ambient temperature in toluene. The amine component (
5) was already known and could easily be prepared.
16 After a simple deacylation with methylamine and subsequent deprotection of the ketone the desired compound
6c was for the first time in our hands. The yield was even better if we allowed amine
5 to react with the
N-unprotected bromoketone
4g, which had been prepared via ketalization of
4d,
N-deacetylation, and a deketalization step in high yields. This sequence yielding
6b and leading from here to
6c proved to be a real shortcut.
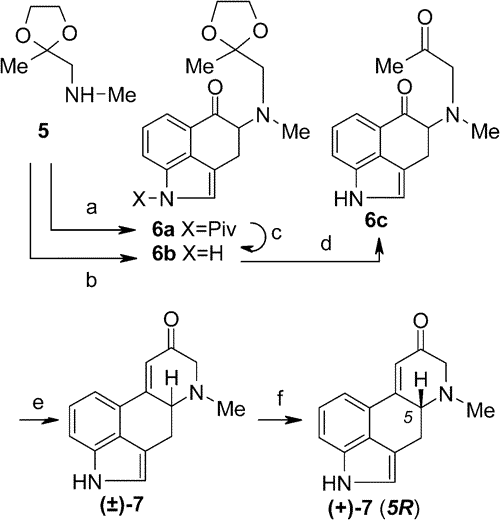 Scheme 3.
Scheme 3. Synthesis of Tetracyclic Ketone [(+)-
7]
a aReagents and conditions: (a)
5 +
4d, toluene, 48 h, rt (35%);
(b)
5 +
4g, THF, 24 h, rt (56%);
(c) MeNH
2, benzene, 10-15 °C, 1 h (80%);
(d) aq HCl (6 M), 35-40 °C, 1 h then
(e)
6c in CHCl
3, LiBr + TEA, 0-5 °C, 12 h (60%, 2 steps);
(f) (-)-dibenzoyl-L-tartaric acid, CH
3CN + H
2O (1:1) (38%).
The ring closure of
6c leading to the unsaturated ketone
717 by intramolecular aldol condensation seems to be an easy task, but with a great number of well-established agents (from potassium
tert-butoxide through super bases to LHMDS) not even a trace of the desired teracyclic compound could be detected. It is worth noting, and not easy to explain, that in the dihydro-indole series this ring closure had been carried out;
16 however, similar intramolecular ring closure of compounds with a sulfone group instead of indole nitrogen also failed. An analogous compound of
6c having an indole
N-tosyl group and an acetyl group on the second nitrogen in place of the methyl group could be closed by KF, but simultaneously isomerization into a naphthalene derivative also occurred.
18 We became successful in performing the reaction by using a LiBr + triethylamine system, which was first used for condensation by Eschenmoser in the case of a different, sulfur-containing compound.
19 LiBr or triethylamine alone were totally ineffective. Likely the LiBr leads to a complementary activation of the two carbonyl groups in the presence of basic amine, since lithium ions have a higher affinity toward oxygen than nitrogen. The function of the amine is purely to abstract the proton in the
alpha-position with respect to the
O-complexed ketone carbonyl. An especially good result was achieved by performing the two consecutive steps (deprotection and ring closure;
6b -->
6c -->
7) without isolation of the intermediate
6c to give a 60% combined yield of crystalline unsaturated ketone
7.
The resolution of
7 was performed with dibenzoyl-tartaric acid. At the same time the optically active ketone was also prepared by degradation of natural lysergic acid,
20 and by comparison the absolute configuration of our synthetic compounds was established.
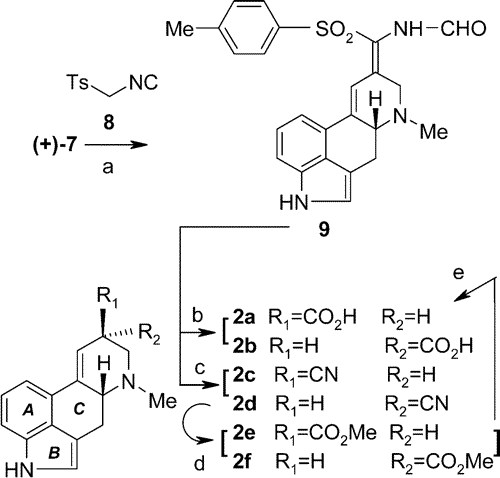 Scheme 4.
Scheme 4. Synthesis of (+)-Lysergic Acid (
2a) from (+)-
7a a Reagents and conditions: (a) (+)-
7 +
8,
t-BuOK, THF +
t-BuOH, 0 °C, 20 min then + H
2O, -5 °C (77%);
(b) aq HCl (2 M), reflux, 30 min (13%);
(c) NaOMe, MeOH, 70-75 °C, 30 min (70%);
(d) HCl/MeOH (6.7 M), 75-80 °C, 45 min (72%);
(e) aq NaOH (5 M), MeOH, 70-80 °C, 2.5 h then aq HCl (6 M) to pH 6.5 (54%).
To proceed, we allowed the optically active
7 to react with the isonitrile derivative
8 in the presence of base
21 to yield the formamide derivative
9, followed by acidic hydrolysis. A mixture of lysergic acid (
2a) and its epimer (
2b) was obtained; after treatment with base almost pure (+)-lysergic acid was isolated as a result of epimerization, although in poor yield.
A much better result was achieved by treating intermediate
9 with base affording a mixture of nitriles (
2c:
2d, 1:1, 70%) and converting the mixture by Pinner reaction into lysergic acid ester diastereomers (a 3:2 mixture of
2e:
2f, 72%). There is no need to separate the two nitriles or esters, since the basic hydrolysis of the mixture of
2e:
2f results in pure (+)-lysergic acid (
2a) through concurrent hydrolysis and epimerization.
22(2) Improving the Efficiency of the Peptide Part Synthesis. Above we described the synthesis of the (+)-lysergic acid component of
alpha-ergocryptine (
1). The synthesis of the peptide part has already been described
23 by a research group from the Sandoz Pharmaceutical Co. Our task was to improve the efficiency, especially the stereoefficiency of the reaction sequence, and to make a scale-up procedure possible.
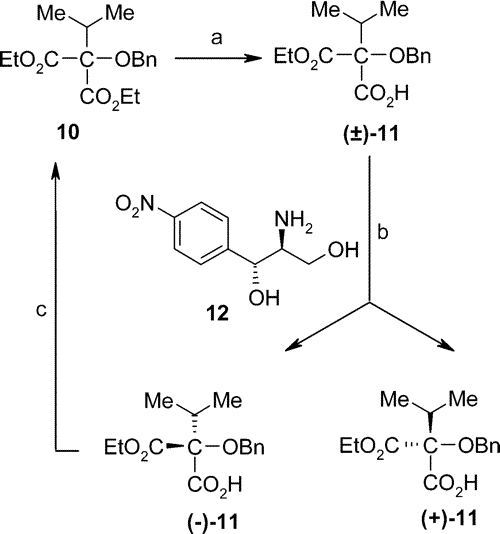
At the outset isopropyl malonic ester was oxidized by benzoyl peroxide. According to the original procedure the excess of the benzoyl peroxide was to be eliminated by charcoal, but following this route we observed explosions in about 20% of the cases. To avoid this danger, Na
2S
2O
3 or NaHSO
3 was successfully used instead of charcoal. The resulting compound was debenzoylated, and the hydroxyl group was protected as the benzyl ether. Partial hydrolysis of diester
10 gave rise to the half ester (±)-
11. In Sandoz's original reaction sequence this acid was resolved by the consecutive application of (-)- and (+)-pseudoephedrine, which process proved to be rather inconvenient and the yield low. Instead of pseudoephedrines we used (+)-1
S,2
S-2-amino-1-(4-nitrophenyl)propan-1,3-diol (
12) for resolution. Compound
12 is the unwanted and thus discarded enantiomer formed during the manufacturing procedure of the antibiotic chloramphenicol.
24 The desired salt of the
R-(+)-isomer [(+)-
11)] crystallized from the solution in excellent yield. Isolation of (+)-
11 has been accomplished by acidic treatment. By this method both (-)-
11 and
12 were recovered easily.
 Scheme 5.
Scheme 5. Modified Resolution of (±)-
11a aReagents and conditions: (a) ref 23;
(b) (±)-
11 +
12, EtOH, rt, 12 h [(+)-
11, 38%; (-)-
11, 39%;
(c) (EtO)
2SO
2, acetone, refl., 3 h (90%).
To make the process even more economic, the unwanted
S-enantiomer [(-)-
11] was esterified with diethyl sulfate to
10. Through this procedure we obtained the original, achiral diester, which we can recycle into the reaction sequence. We may call this manipulation
dechiralization.
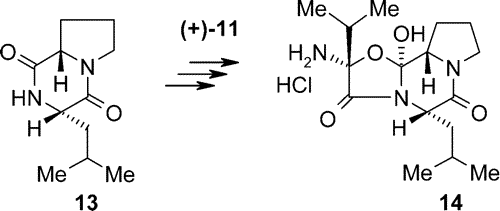
The so-called aminocyclol hydrochloride (
14), the partner needed for coupling with (+)-lysergic acid, was prepared from Z-protected proline. The proline derivative was treated with L-leucine methyl ester
p-tolyl sulfonate salt using the mixed anhydride (chloroformic acid ester) method. After deprotection by hydrogenolysis followed by heating, the L-prolyl-L-leucyl lactam (
13) was isolated in good yield.
The malonic acid derivative [(+)-
11] was transformed to the acid chloride and allowed to react with lactam
13, then deprotected by hydrogenolysis, and the resulting cyclolester hydrolyzed to the so-called cyclolcarboxylic acid. After several steps
14 was btained.
 Scheme 6
Scheme 6Several methods were tried for coupling lysergic acid (
2a) with the peptide part (
14). The most practical route was found by using lysergic acid trifluoroacetate, which was allowed to react with PCl
5. The reaction conditions (temperature, the excess of reagent) are critical. The approximate amount (80%) of acyl chloride in the obtained reaction mixture was estimated by IR spectra. By reacting the suspension of the aminocyclol hydrochloride in methylene chloride with lysergic acid chloride hydrochloride
25 at -12 °C in the presence of pyridine,
alpha-ergocryptine (
1) was isolated in 41% yield (as its phosphate salt).
26 In addition to its diastereomer
alpha-ergocryptinine (
15) was obtained (31%) after chromatographic workup.
23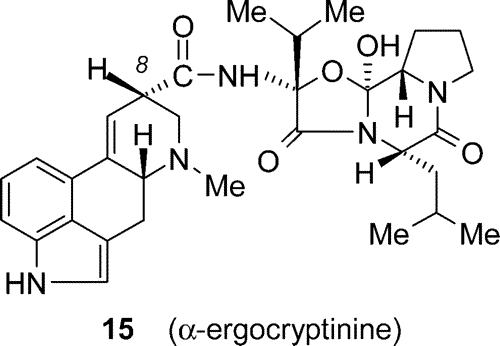 Scheme 7
Scheme 7Since a thermodynamic equilibrium exists between the two stereoisomers in favor of
1 to
15 (3:1) in boiling methanol or in other solvents, in principle there is a possibility to transform
15 into
1 in preparative scale. This aspect, however, was not closely investigated.

