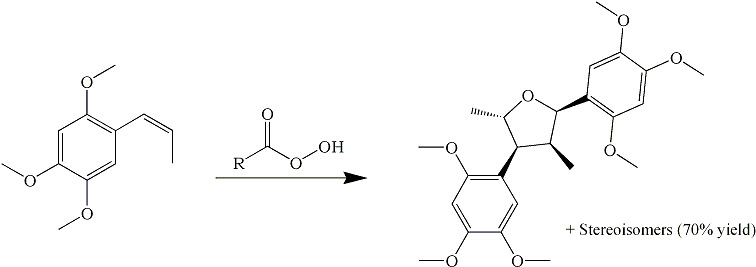
I have finally found an article which deals with the precise reasons why asarone cannot undergo the performic oxidation to form a glycol, which then can be dehydrated and rearranged to asarone ketone (TMP2P).
According to
Tetrahedron 42, 523-528 (1986), attempted oxidation of

-asarone with peracetic acid at room temp gave the above heavily substituted tetrahydrofuran (a naturally occuring neolignan, called Magnosalicin) in 16% yield along with its three other stereoisomers (same structure (2,4-dimethyl-3,5-bis(2',4',5'-trimethoxyphenyl)-tetrahydrofuran) but with the substituents pointing in other directions), the total yield being about 70%.
Magnosalicin is a red solid, while the other isomers are oils. This may account for the fact that the reaction mixture usually is thick and red after attempted performic or peracid oxidation of asarone.
The reaction mechanism is proposed to be the epoxide opening up under acidic conditions to give TMP2Pol with a carbocation in the benzylic position (which is stabilized because of the electron-donating methoxy groups on the aromatic ring). This carbocation then adds to the 2-position on the side-chain of an unreacted asarone molecule, forming a carbon-carbon bond, as well as a new carbocation in the benzylic position of the attacked asarone molecule. This immediately adds to the alcohol and the tetrahydrofuran ring is formed.
There has been reports of people being able to oxidize asarone using buffered peracetic acid, and this is due to the formed epoxide not opening up under the reaction conditions, as the mixture is much closer to neutral than in the unbuffered reaction. The epoxide not opening up means that there will be no reactive carbocation formed, which could otherwise dimerize.
The question is now - is this a side reaction in the oxidation of isosafrole, and can it be prevented? There seems to be another paper on this topic which can shed some light on this, in which they on an industrial scale oxidize anethole with peracetic acid, isolating the corresponding tetrahydrofuran in 5-10% yield (bp 135-160°C/0.15mmHg) -
Pharmazie 34, 22 (1979)
(https://www.thevespiary.org/rhodium/Rhodium/pdf/anethol.peracetic.pdf) As the aromatic ring of isosafrole is of intermediate electron richness, being somewhere between asarone and anethole, there is every reason to believe that in the unbuffered peracid oxidation of isosafrole, 2,4-dimethyl-3,5-bis(3,4-methylenedioxyphenyl)-tetrahydrofuran is formed to an extent of between 10% and 40% yield (higher than anethole, but with a "roof" of ~40%, because evidently people have gotten yields of at least 60% using that route).

