The long awaited Glycine to Methylamine reference
As i'm sure many of you know there has been sporatic talks of the conversion of glycine to methylamine throughout the history of the hive. As many of you know glycine is about the simplest form of amino acid, and the only amino acid with no stereoisomers. It is easily obtainable and is therefore a viable and expedient method of obtaining methylamine for bees with a knowledge of organic chemistry.
There was much talk of heating glycine with a base (as it turns out it seems that water is adequate), and a large following who considered the reaction very dirty, experiencing polymerization, byproducts, and after gassing recieving impurities in the salt.
Everyone could remember reading something about it years ago but no one could seem to come through with anything that explained the process in any detail for those who would like to optimize the reaction to obtain methylamine from this convienient method. The article is not in direct reference to methylamine production, but explains the mechanics fairly well and will certainly help better the understanding of the reaction for it's optimization.
Herein is the recently published explanation of the migration of the methyl groups.
Migration of Methyl Groups between Aliphatic Amines in WaterBrian P. Callahan and Richard WolfendenDepartment of Biochemistry and Biophysics,
University of North Carolina, Chapel Hill, NC
Abstract : Glycine undergoes spontaneous decarboxylation in dilute aqueous solution at elevated temperatures to form methylamine. During that process, we noticed the apperance of dimethylamine and trimethylamine in smaller amounts that increased gradually with time. These observations suggested the existence of disproportionation reactions of methylamines in water, for which there appears to be no direct precedent in the literature. Every member of the methylamine series is found to yield other members of the methylamine series. When the total concentraion of amine was held constant and the rate of reaction was examined as a function of changing pH using the amine itself as the buffer, the initial rate of appearance of the products was found to reach a maximum when the conjugate acid and the conjugate base were present at equivalent concentrations. Near this equivalence point, the rate of reaction varied with pH as expected for a second-order reaction between the protonated and the unprotonated species. Under similar conditions, methyl groups were also found to migrate between the nitrogen atoms of N,N-dimethyl-1,3-propanediamine in a first-order process. With dimethylamine as a common acceptor, trimethlsulfonium ion was found to be ~10
4-flod more reactive that the tetramethylammonium ion at ambient temperature.
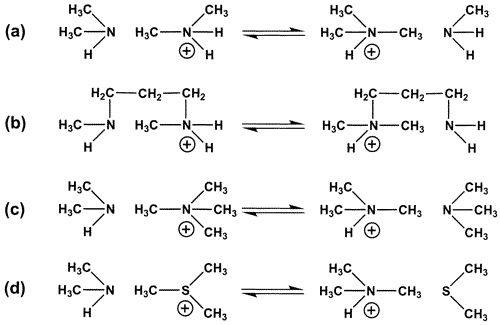
Glycine undergoes spontaneous decarboxylation in dilute aqueous solution at elevated temperatures to form methylamine. During that process, we noticed the apperance of dimethylamine and trimethylamine in smaller amounts that increased gradually with time. These observations suggested the existence of disproportionation reactions of methylamines in water, for which there appears to be no direct precedent in the literature. Here, we show that methyl groups migrate between aliphatic amines when they are incubated with their conjugate acids at elevated temperatures and that competition by water as a methyl acceptor is negligible. Half-titrated dimethylamine (0.02 M), for example, yields trimethylamine and methylamine in equal amounts (Scheme 1a), with no appearance of methanol. In addition to the apparent novelty of these reactions, these results are of interest in relation to the mechanism of methyl transfer in biological systems.
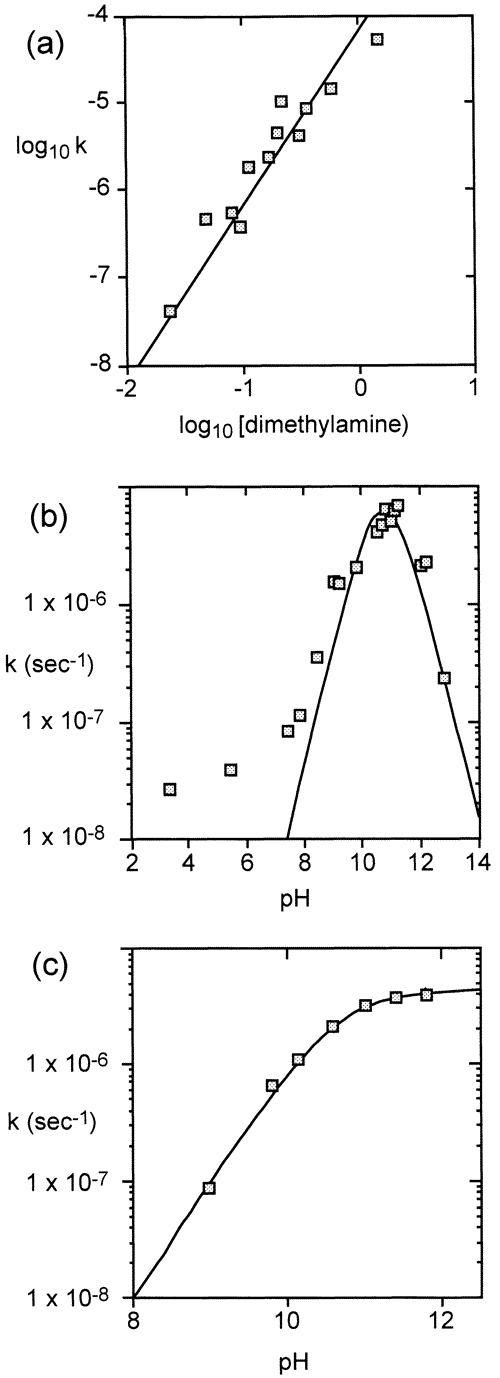
To determine the rates of methyl transfer between amines, we incubated aqueous amines, half-titrated with HCl, for various time intervals at high temperatures in stainless-steel bombs lined with PTFE. After cooling, the reaction mixtures were removed and analyzed by proton NMR, using added pyrazine as an integration standard. Every member of the methlamine series was found to yeild other members of the methylamine series. AT each of a series of temperatures, the rate of disappearance of the starting material followed satisfactory first order kinetics and yielded a linear Arrhenius plot when the logarithm of the initial rate of reaction was plotted as a function of the reciprocal of the absolute temperature. When the ratio of the protonated to the unprotonated species of mono-, di-, or trimethylamine was held constant but the total concentration of that amine varied, the initial rate of disappearance of the starting material was found to vary in proportion to the square of its concentration, as expected for a bimolecular reaction involving two molecules of amine (see for example Figure 1a).
When the total concentration of amine was held constant, and the rate of reaction was examined as a function of changing pH using the amine itself as the buffer, the initial rate of appearance of the products was found to reach a maximum when the conjugate acid and the conjugate base were present at equivalent concentrations. Near this equivalence point, the rate of reaction varied with pH as expected for a second-order reaction between the protonated and the unprotonated species, as shown by the solid line in Figure 1a. At pH values much below this equivalence point, product formation continued at a much slower rate (Figure 1b). The persistence of reactivity at low pH, which has also been reported for other transalkylation reactions, suggests that water may be acting as a general base catalyst.
In experiments in which the tetramethylammonium ion (Me4N+) was employed as the methyl donor and dimethylamine was the acceptor, trimethylamine was formed at an initial rate proportional to the concentrations of each of these two reactants. When this reaction was followed as a function of increasing pH, using dimethylamine itself as a buffer,3 its initial rate of disappearance became half-maximal at the pH value where dimethylamine is half-converted to its uncharged form, approaching a constant value when dimethylamine is fully converted to its uncharged form but Me4N+ retains its positive charge (Figure 1c). Activation parameters based on the resulting Arrhenius plots are shown in Table 1.
Table 1.
Extrapolated Rate Constants and Activation Parameters
from Arrhenius Plots, Based on 10 or More Rate Constants
Obtained over a Range >50 C, with Estimated Errors in ^H and
T^S of ±1.5 kcal/mol
a. k (25 C) (M-1 s-1)
b. H, kcal/mol
c. TS, kcal/mol
dimethylamine + dimethylammonium
a. 4 × 10-13
b. 25.9
c. -8.5
N,N'-dimethyl-1,3-propanediamine (s-1)
a. 5 × 10-12
b. 30.4
c. -2.5
dimethylamine + tetramethylammonium
a. 1.9 × 10-12
b. 30.1
c. -3.4
dimethylamine + trimethylsulfonium
a. 1.5 × 10-8
b. 22.2
c. -5.9
Figure 1 (a) Initial rate of conversion of half-titrated dimethylamine to methylamine and trimethylamine at 226 C, plotted as a logarithmic function of changing initial concentration of dimethylamine. The line (slope = 2) is calculated for a reaction that is of the second order with respect to the total concentration of amine (Scheme 1a). (b) Initial rate of the same reaction at 226 C, at a fixed total concentration of dimethylamine (0.5 M), plotted as a function of changing pH. The line represents the behavior expected for the reaction shown in Scheme 1a. (c) Initial rate of reaction of dimethylamine (0.6 M) with tetramethylammonium chloride (0.25 M) at 171 C, plotted as a function of effective pH. The line represents the behavior expected for the reaction shown in Scheme 1c.