I haven't read whole this thread but I guess you want to go to 3,4,5 pattern with this starting material. You can formylate this compound in 100% street fashion, getting syringaldehyde, the yields are pretty bad, it is IMO not a good route, but you can alternatively go from vanillin of course also OTC;
Organic Syntheses, CV 4, 866
(
http://www.orgsyn.org/orgsyn/prep.asp?prep=cv4p0866)
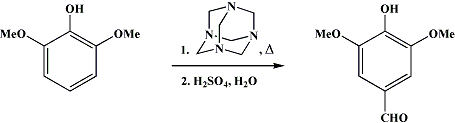
A well-stirred (Note 1) and (Note 2) mixture of 740 ml. of glycerol and 216 g. of boric acid, in a 2-l. three-necked round-bottomed flask fitted with a thermometer and a condenser for downward distillation, is dehydrated by heating in an oil bath to exactly 170°. This temperature is maintained for 30 minutes and then allowed to drop. When the temperature has fallen to 150°, a mixture of 154 g. (1 mole) of pyrogallol-1,3-dimethyl ether and 154 g. (1.1 moles) of hexamethylenetetramine (Note 3) is added as rapidly as possible through the neck holding the thermometer. The temperature drops to approximately 125°. Rapid heating is immediately started but is slowed down when the temperature begins to reach 145° and stopped at 148°. The reaction must be watched and controlled very carefully when this temperature is reached, since the reaction becomes exothermic at this point (Note 4), (Note 5), and (Note 6). The temperature is maintained at 150–160° for approximately 6 minutes (Note 7). At the end of this reaction time the mixture is cooled to 110° as rapidly as possible (Note 6) and (Note

, and a previously prepared solution of 184 ml. of concentrated sulfuric acid in 620 ml. of water is added to the reaction mixture. After being stirred for 1 hour, the mixture is cooled to 25° in an ice bath. The boric acid, which separates from the solution, is removed by filtration (Note 9) and washed free of mother liquor with 400 ml. of water. The filtrate and washings are combined and extracted with three 500-ml. portions of chloroform (Note 10), (Note 11), and (Note 12).
The chloroform solution is then extracted with a filtered solution of 180 g. of sodium bisulfite in 720 ml. of water (Note 13) by stirring rapidly with a Hershberg stirrer for 1 hour. The separated bisulfite solution is washed twice with chloroform, filtered, and acidified in a hood with a solution of 55 ml. of concentrated sulfuric acid in 55 ml. of water. After careful heating on a steam bath for a short time, air is bubbled through the hot solution until all the sulfur dioxide has been expelled. The product, which separates as a mixture of crystals and oil, readily solidifies upon cooling (Note 14). The syringic aldehyde is collected by filtration, washed with cold water, and dried in an oven at 40° to give 62.5–66 g. of light-tan material, melting at 110.5–111°, which still contains a small amount of foreign material that does not melt at 300°. Recrystallization of the crude product from aqueous methanol using 30 ml. of water and 3 ml. of methanol for each 10 g. of aldehyde gives 56–59 g. (31–32%) of product melting clear at 111–112° (uncor.). A second extraction of the chloroform solution with a filtered solution of 60 g. of sodium bisulfite in 240 ml. of water gives an additional 3–4 g. of product.
I would like to see this formylation on phloroglucinol.

