Synthesis of homogentisic acid by carbonylation
Chalasani S N Prasad & Srinivas R Adapa
Indian Journal of Chemistry Vol. 31B, pp. 626-627 (1992)
(https://www.thevespiary.org/rhodium/Rhodium/pdf/homogentisic.pdf)Homogentisic acid is an important intermediate in the overall catabolic process of phenylalanine and tyrosine
[1], responsible for the formation of pigment melanin. Several reports, on the synthesis of homogentisic acid, have appeared by virtue of its biological importance. All involve a multistep sequence starting either from p-benzoquinone or its derivatives
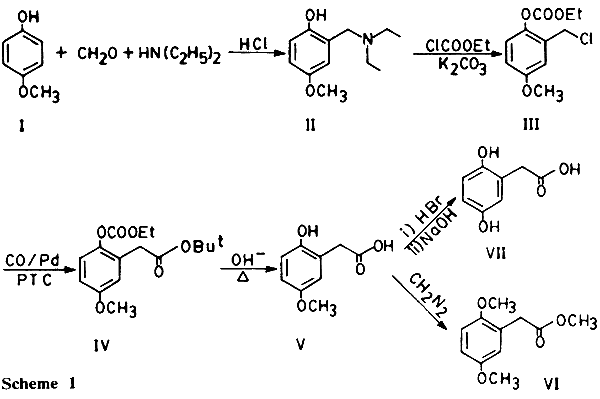
[2,3,4]. Its synthesis by carbonylation appears not to have been described so far. In this note we report its synthesis via the preparation of 2,5-dihydroxybenzyl chloride derivative from hydroquinone monomethyl ether followed by carbonylation and hydrolysis. A Mannich reaction using formaldehyde and diethyl amine on hydroquinone monomethyl ether (
I) results in the formation of the derivative (
II), which on treatment with cathyl chloride (ethyl chloroformate) leads to O-carbethoxy-5-methoxybenzyl chloride (
III) which on carbonylation under mild conditions using PTC-PdL
2Cl
2 as catalyst
[5] forms the corresponding t-butyl phenyl acetate derivative (
IV).
IV on hydrolysis under reflux with NaOH results in the formation of 2-hydroxy-5-methoxyphenylacetic acid (
V). Demethylation and methylation of
V gives homogentisic acid (
VII) and its methyl ester dimethyl ether (
VI) respectively (Scheme 1).
 Experimental2-Hydroxy-5-methoxy-N,N-diethylbenzylamine (II)
Experimental2-Hydroxy-5-methoxy-N,N-diethylbenzylamine (II)To a mixture of p-methoxyphenol (32 g; 30 mmol) and diethylamine (130 ml; 125 mmol) under nitrogen atmosphere at 70°C was added paraformaldehyde (6 g) while stirring and the mixture refluxed for 6 hr. The mixture was cooled to 50°C and diethyl amine removed on a rotary evaporator. The mixture was cooled to room temperature and 10% aqueous HCl added to make the solution acidic. The solution was extracted with ether to remove unreacted starting material. The aqueous HCl portion was basified with 10% liquor ammonia and extracted with dichloromethane. The combined dichloromethane phase was dried (K
2CO
3) and dichloromethane removed on a rotary evaporator. The resulting product was subjected to Kugel Rohr distillation to give 2,5-dihydroxyphenyl derivative (
II), yield 52 g (85%); bp 90°C/5mmHg.
2-Oxo-carbethoxy-5-methoxybenzyl chloride(III)To a solution of benzylamine (
II) (42 g, 20 mmol) in chloroform (50 ml) was added anhydrous potassium carbonate (12.5 g; 90 mmol) and treated slowly with ethyl chloroformate (20 ml, 19 mmol, acid free) in 2 hr at 0°C while stirring. The mixture was stirred at 0°C for another 2 h and diluted with CHCl
3/water (1:1, 100 ml). The combined organic phase was washed with 1% HCl (2x25 ml) followed by water (4x5 ml) and dried (CaCl
2). The solvent was evaporated and the residue purified by bulb to bulb distillation to give
III, yield 39 g (80%), bp 85°C/2mmHg.
2-Oxo-carbethoxy-5-methoxy-t-butyl phenyl acetate (IV)Compound
IV was prepared by carbonylation of
III (4.5 g, 20 mmol) by the literature method
[5] and purified by bulb to bulb distillation
IV; yield 2.5 g (40%); bp 78°C/2mmHg.
2-Hydroxy-5-methoxyphenylacetic acid (V)To a solution of compound
IV (3.1 g, 10 mmol) in ethanol (30 ml), aqueous sodium hydroxide (0.5 N, 30 ml) was added and the mixture refluxed for 6 hr. The mixture was cooled, diluted with water (60 ml) and extracted with ether (3x50 ml). The aqueous portion was cooled, acidified with conc. HCl and extracted with ether (3x50 ml) and dried (Na
2SO
4). Evaporation of the solvent gave the crude product which was crystallised from petroleum ether (40-60°) gave pure
V, yield 0.55g (30%), mp 82°C.
Methyl ester of homogentisic acid dimethyl ether (VI)To a solution of
V (91 mg, 0.05 mmol) in ether, distilled diazomethane was added while stirring at 0-5°C. The mixture was stirred for 2 hr at 0-5°C and the solvent removed in rotary evaporator to give the crude ester which was purified by recrystallisation from pet. ether (40-60°) to give
VI; yield 60 mg (60%), mp 43°C (lit
[4] mp 45°C).
References[1] Knox W C & Le May-Knox M,
Biochem. J. 49, 686 (1951); LaDU B N & Zannani V G,
J. Biol. Chem. 217, 777 (1955).
[2] DeForrest Abbott (Jr) L & Smith J D,
J. Biol. Chem., 179, 365 (1949)[3] Bostock S B & Renfrew A H,
Synthesis, 66 (1978)[4] Bloomer 1 L & Damodaran K M,
Synthesis, 111 (1980)[5] Adapa S R & Prasad C S N,
J. Chem. Soc. Perkin. Trans. 1, 1706 (1989)

