Some Extensions of von Braun (BrCN) Reaction on Organic Bases. Part I.Salimuzzaman Siddiqui and Bina S. SiddiquiZ. Naturforsch. 35B, 1049-1052 (1980)
(
https://www.thevespiary.org/rhodium/Rhodium/pdf/von.braun.cnbr-1.pdf)
AbstractSome extensions of von Braun cyanogen bromide reaction have been undertaken on conessine, isoconessine and two simpler bases, dimethyl ?-naphthyl amino and diethylamine. The monocyanamides of conessine and isoconessine yielded acid amides, amino-derivatives (diamines) and guanido derivatives on careful hydrolysis, reduction and treatment with ammonia, respectively. The simpler bases also formed the acid amides and diamines but failed to give the guanido derivatives under the conditions employed for conessine series. Diamines of all those bases yielded carbinol amines on reaction with nitrous acid.
____ ___ __ _
Some Extensions of von Braun (BrCN) Reaction on Organic Bases. Part II.Malik, Abdul; Afza, Nighat; Siddiqui, SalimuzzamanZ.Naturforsch.B Anorg.Chem.Org.Chem. 37(4), 512-518 (1982)
(
https://www.thevespiary.org/rhodium/Rhodium/pdf/von.braun.cnbr-2.pdf)
AbstractExtensions of von Braun Cyanogen bromide reaction on Ephedra alkaloids and simpler bases have resulted in synthesis of substituted oxazolidines and a whole series of nitrogen analogues of ephedrine, desoxyephedrine and simpler amines. The general applicability and limitations of such extension of the reaction are also discussed.
The present work deals with the cyanamides of ephedrine (1), O-acetyl ephedrine (2) and desoxyephedrine (3) which were obtained as oily liquids through the action of BrCN. Attempts were made to obtain from ephedrine cyanamide an N-amide, introducing a urea moiety in the molecule, through careful partial hydrolysis with 20% hydrochloric acid. However, the resulting uniform oily product did not show the presence of amide group in the IR and NMR spectra. Ir could be identified through physical and chemical characterization, and spectral studies as 2-imino-3,4-dimethyl-5-phenyl oxazolidine (4) (yield 1 g; 100%). The protonation of the nitrile nitrogen promotes nucleophilic attack from the hydroxyl group resulting in a cyclized product. This constituted a better method of preparation than that described by Fodor et al.
[G. Fodor K. Koezka, and L. Szekeres, Acta Chim. Acad. Sci. Hung. 1, 377 (1951)] which involves a rather cumbersome three steps process resulting apparently in low yield.
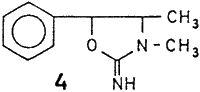
Reduction of 4 with zinc and hydrochloric acid furnished colourless glistening rods of 2-amino-3,4-dimethyl-5-phenyloxazolidine (5), mp 99°C (yield 1.01 g; 95%). Further, the amino derivative (5) has yielded 2-hydroxy-3,4-dimethyl-5-phenyl oxazolidine (6) as colourless fluffy needles, mp 92°C (yield 0.84 g; 83.6%) through careful reaction with nitrous acid. Both these products were hitherto unreported in literature and their structures as shown below, were confirmed through physical and chemical characterization along with spectral data.
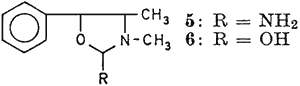
In order to prevent hydroxyl induced cyclization ephedrine was converted into desoxy ephedrine by the method of Schmidt
[Arch. Pharm. 251, 320 (1913)]. As a result of extension of von Braun reaction on desoxy ephedrine it has been possible to obtain from desoxy ephedrine cyanamide (3) an N-amide (

(yield 0.98 g; 89%) through partial acid hydrolysis. On the other hand, through reduction with zinc and hydrochloric acid, a diamine (9) has been obtained (yield 0.777g; 76%). Further, the diamine has yielded a carbinol amine (10) through reaction with nitrous acid (yield 0.613g; 61%). The structure of these products as shown below, have been arrived at through spectral data and the formation of various derivatives, which are described in the experimental.
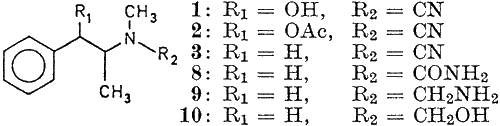 ExperimentalCyanamide derivatives of Ephedra alkaloids
ExperimentalCyanamide derivatives of Ephedra alkaloids The cyanamide of ephedrine and desoxyephedrine were prepared by treating their chloroform solution with freshly prepared ethereal solution of BrCN, under good cooling and mechanical stirring for 1 h. The crystalline hydrobromide formed in the reaction was filtered and washed with ether. On removal of the solvent from filtrate and washings, after washing and drying yielded cyanamide derivatives as colourless viscous liquids. The similar work up with N-methyl-O-acetyl ephedrine provided O-acetyl ephedrine cyanamide.
Substituted Oxazolidines2-Imino-3,4-dimethyl-5-phenyloxazolidine1 g ephedrine cyanamide was suspended in 10 ml of 20% hydrochloric acid and mechanically stirred with occasional warming till a clear solution was obtained. The reaction mixture was basified with ammonia and extracted out with ethyl acetate. The basic oily residue left on removal of the solvent was taken up in ether and treated with ethereal HCl. The resulting hydrochloride crystallized from methanol in shining needles mp 235°C.
2-amino-3,4-dimethyl-5-phenyl oxazolidine 1 g imino derivative referred to above, was heated with zinc dust and 10% aqueous HCI on water bath for half an hour the solution was filtered and basified with ammonia after prior addition of ammonium chloride. The liberated base was extracted out with ethyl acetate and after usual work up crystallized out from methanol as colourless glistening rods mp 99°C.
2-Hydroxy-3,4-dimethyl-5-phenyl oxazolidineTo a solution of 1 g amino base in 10% hydrochloric acid was added a 10% cold aqueous solution of sodium nitrite (1.2 mole) and after 15 min, the reaction mixture was extracted out with ethyl acetate. The darkish ethyl acetate layer was treated with ether and petroleum ether which threw out the resinous impurities. The nearly colourless solution gave a light yellow residue which crystallized from methanol as fluffy needles, mp 92°C.
Ephedrine-N-iminoethyl ether1 g of ephedrine cyanamide was taken up in 10 ml absolute alcohol and to it was added 100 mg of sodium metal. After half an hour the reaction mixture was extracted out with ethyl acetate with the addition of saline. On working up the ethyl acetate extract N-imino ethyl ether derivative was obtained as colourless rectangular plates from methanol which melted at 105°C (yielded 0.782 g, 63%).
Derivatives of desoxy ephedrineDesoxy ephedrine urea (2-methylamino-1-phenylpropane urea): Ephedrine cyanamide (1 g) was taken in 20% aqueous hydrochloric acid and stirred at 60°C for 1 h when it went into solution. Stirring was continued for further 10 min after which the solution was cooled, basified with ammonia and extracted out with ethyl acetate. The product crystallized out from methanol as colourless shining plates mp 120°C.
N-Aminomethyl desoxyephedrineDesoxyephedrine cyanamide (1 g) was taken in 15% aqueous HCl and heated with zinc dust on water bath till it went into solution. The heating was continued for about half an hour after which the unreacted zinc was filtered off and the filtrate ammoniated after adding ammonium chloride. The product crystallized out from methanol as colourless rectangular plates, mp 135°C.
N-Hydroxy methyl desoxyephedrine1 g N-amino methyl derivative was taken in 10% aqueous HCl and 1.2 mole of sodium nitrite in cold aqueous solution was added on to it. The N-hydroxymethyl derivative was obtained on usual work up as colourless shining needles from methanol mp 120°C.

