J. Chem. Research (S), 2003, 580-583
(
http://lego.chemistry.tripod.com/Journals/tropinone.pdf)
No DOI foundTheoretical study on the mechanism of Robinson’s synthesis of tropinoneNityagopal Mondal, Sannyasi Charan Mandal, Gourab Kanti Das and Sarbananda Mukherjee
Department of Chemistry, Visva-Bharati, Santiniketan-731235, India
Abstract: Ab initio quantum mechanical calculation reveals that the first Mannich reaction in Robinson’s tropinone synthesis involves both carbon–carbon bond formation and water elimination, which is followed by tautomerisation and a second Mannich reaction to form the protonated tropinone.
Keywords: tropinone, aldol condensation, transition structure
Synthesis of the alkaloid (±)tropinone, reported as early as 1917 by Robinson, is still an exciting example of total synthesis.
1 In this synthesis Robinson utilised the reaction between one molecule of succinaldehyde, methylamine and acetone (or its dicarboxylic acid derivative) in a simple one-pot procedure (Scheme 1).
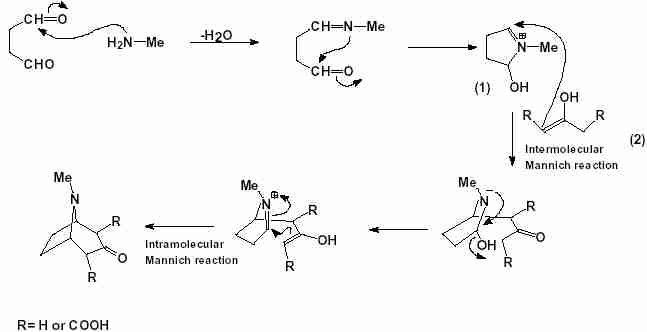 Scheme 1
Scheme 1 Total synthesis of (±)tropinone by Robinson.
Two consecutive Mannich reactions for C–C bond formation are involved in this synthesis. The first one is an intermolecular reaction between
1 (a pyrrolidine derivative) and
2 (enol form of acetone or its dicarboxylic acid derivative) and the second one occurs in an intramolecular fashion. It has been believed that after the first Mannich reaction elimination of a water and tautomerisation of the product molecule occurs to generate the reactant for the second Mannich reaction. The mechanism of the Mannich reaction is similar to that of the aldol condensation. Though a number of theoretical works on simple, metal-catalysed
2 or imine-catalysed aldol condensations
3 are found in the literature, no such theoretical study has been made of the synthesis of tropinone. In this short report we present our quantum chemical study on this reaction. In contrast to the accepted mechanism, in which the first Mannich reaction and iminium ion formation is considered as a separate step, our study on the gas phase reaction reveals that the most favourable transition structure for the first step is also responsible for the water elimination (i.e. the two processes occur in a single step). Tautomerisation and the second Mannich reaction may then take place for the formation of the basic skeleton of the target molecule.
Method of calculation: In this investigation all structures were optimised using a HF/6-31G* basis set as implemented in GAMESS software
4. All gas phase minima and transition structures (transition states or TS) were characterised by frequency analysis. For confirming the stability of a particular conformation among various ones, single point energy calculation using density functional theory (DFT) has also been used.
Results and discussionWe started our search by finding various possible TSs generated by the reaction between reactant
1 (a pyrolidine derivative) and
2 (enol form of acetone). In the first step, nucleophilic attack by reactant
1 on
2 takes place as shown in the mechanistic diagram of Scheme 1. In reactant
1, presence of an asymmetric and a prochiral centre, makes two diastereotopic faces (
re and
si) (Fig.1) for the attack of the second reactant
2. Reactant
2 also possesses two enantiotopic faces.
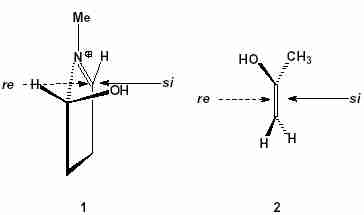 Fig.1
Fig.1 Diastereotopic faces of reactant
1 (in the
R configuration) and enantiotopic faces of reactant
2.
Hence in each configuration (R or S) of
1, nucleophilic attack by
2 results in four diastereomeric transition states. Previous theoretical reports on simple metal-catalysed
2 or the aminecatalysed
3 aldol condensations show that C–C bond formation generally takes place via transition structures with staggered conformations. Our search also results in three such conformational isomers in the staggered form for each diastereomeric TS. Table 1 shows the energies of all the TSs obtained from the
R configuration of reactant
1. Each TS may exist in its enantiomeric form and they are of the same energy state in the achiral enviroment. The relative energies (calculated by HF/6-31G* and B3LYP/6-31G*) show that the most favourable transition structure TS-1 is formed from the reactant
1 by the attack of
2 from the
si face in the –
sc conformation.
Table 1 Energies of the diastereomeric transition structures in the first Mannich reaction between
1 and
2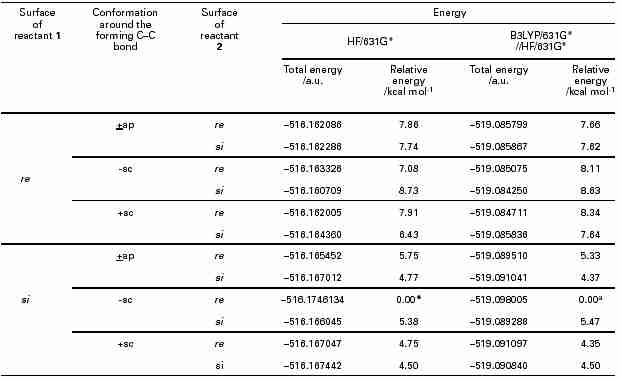 a
a Favourable TS with lowest energy. Relative energies are calculated with respect to this TS.
Figure 2 shows the model of TS-1.
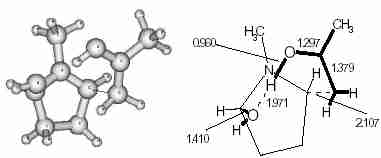 Fig.2
Fig.2 Transition state (TS-1) for the first Mannich reaction (intermolecular).
The high stability of the favourable TS relative to others is due to the hydrogen bond between the enolic hydrogen and the hydroxyl group attached to the pyrrolidine moiety. An IRC calculation using the standard algorithm (steepest descent method) fails to give reactants or the product from the TS. We followed the pseudo reaction co-ordinate
5 by fixing the forming C–C bond distance and allowing the rest of the geometry to optimise. The fixed parameter (C–C bond distance) was then stepped towards the normal C–C bond distance. At each step the structure was reoptimised. It was found that during the formation of the C–C bond the hydroxyl group of the enol part in reactant
2 is deprotonated and the leaving proton combines with the hydroxyl group of the pyrrolidine part in reactant
1 to form a detached water molecule (Fig.3). Hence TS-1 not only directs the C–C bond formation but is also responsible for the elimination of water to generate the product (P1)
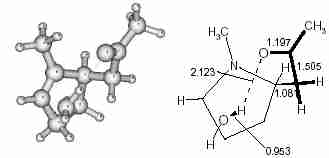 Fig. 3
Fig. 3 Geometry of P1, the product obtained from TS1.
This observation allows us to draw the first step as a cyclic process as shown in Scheme 2.
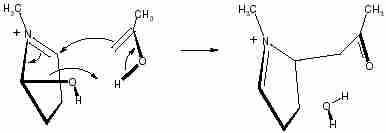 Scheme 2
Scheme 2The second step involves the tautomerisation. We have investigated this process as a water-assisted step in which an external water molecule transfers one hydrogen atom from the carbon to the oxygen atom of the reactant by a relay mechanism. The TS (TS-2) is shown in Fig 4. IRC calculation confirms the formation of the enol from the keto structure through TS-2. Although we have used a single water molecule as catalyst, more than one molecule may be involved in tautomerisation and that may also reduce the activation energy of the process.
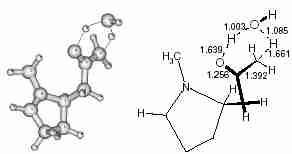 Fig. 4
Fig. 4 Transition state of the solvent (water) assisted tautomerisation (TS-2).
The last step involves the formation of another C–C bond and the corresponding TS (TS-3) is shown in Fig 5.
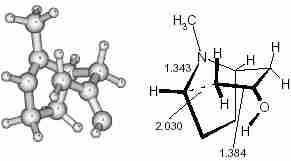 Fig. 5
Fig. 5 Transition state (TS-3) of the second Mannich reaction (intramolecular) for C–C bond formation.
The model shows that a six membered piperidine moiety is generated during the formation of the second C–C bond and the overall configuration of this forming ring is the chair form. The methyl group, attached to the nitrogen, is in an axial position and the oxygen of the hydroxyl group is almost coplanar with its adjacent three carbon atoms. IRC calculation shows that the reactant, obtained from this TS, is a rotomer of the product obtained in the tautomerisation step. So after tautomerisation the system should have a conformational change by the rotation about the C–C bond, formed in the first step. Figure 6 shows the energy profile of the overall process of tropinone synthesis. The authors are thankful to UGC, New Delhi, India for providing financial support for this research work.
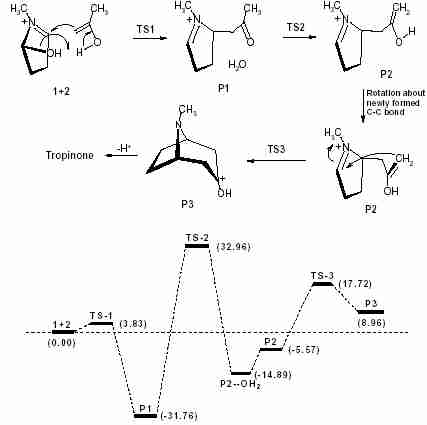 Fig.6
Fig.6 The overall process along with the energy profile diagram (kcal mol
-1).
Received 20 January 2003; accepted 10 June 2003
References1 (a) K.C. Nicolaou, D. Vourloumis, N. Winssinger and P.S. Baran, Angew. Chem. Int. Ed., 2000, 39, 44; (b) I. Fleming, Selected Organic Syntheses, Wiley, New York, 1973; (c) R. Robinson. J. Chem. Soc., 1917, 111, 762; (d) Lousnasmaa, The Alkaloids, ed. A. Brossi, Academic Press, New York,1988,33, 1; (e) E. Leete and S.H. Kim, J. Chem. Soc., Chem. Commun., 1989, 1899.
2 K.W. Henderson, A.E. Dorigo, Q. –Y. Liu, P.G. Williard, P.V.R. Schleyer and P.R. Bernstein, J. Am. Chem. Soc., 1996, 118, 1339; (b) Y. Li, M.N. Paddon-Row and K.N. Houk, J. Org. Chem., 1990, 55, 481; (c) A. Bernardi, A.M. Capelli and C. Gennari, J. Org. Chem., 1990, 55, 3576.
3 L. Hoang, s. Bahmanyar, K.N. Houk and B. List, J. Am. Chem. Soc., 2003, 125, 16; (b) S. Bahmanyar and K.N. Houk, J. Am. Chem. Soc. 2001, 123, 12911; (c) S. Bahmanyar and K.N. Houk, J. Am. Chem. Soc., 2001, 123, 11273.
4 M.W. Schmidt, K.K. Baldridge, J.A. Boatz, S.T. Elbert, M.S. Gordon, J.J. Jensen, S. Koseki, N. Matsunaga, K.A. Nguyen, S. Su, T.L. Windus, M. Dupuis and J.A. Montgomery, J. Comput. Chem., 1993, 14, 1347.
5 D.C. Young, in Computational Chemistry, Wiley-Interscience, John Wiley & Sons, Inc., 2001.
See also:
Post 338869
(Tricky: "Robinson's tropinone: improving method!", Novel Discourse)
