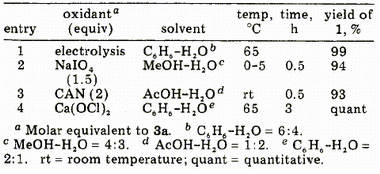
Table I. Electrochemical and Chemical Oxidations of Isosafrole Glycol 3a
Experimental SectionThe
1H and
13C NMR spectra were measured in CDCl
3 with Me
4Si as an internal standard on a JEOL FX-9OQ spectrometer. The IR spectra were obtained with a JASCO IRA-1 spectrometer.
Electrolysis of Isosafrole (2). Preparation of 1-[3,4-(Methylenedioxy)phenyl]propane-1,2-diol (3a).A mixture of
2 (100 mg, 0.62 mmol) and NaBr (192 mg, 1.9 mmol) dissolved in MeCN (7 mL) and H
2O (3 mL) was electrolyzed in a beaker-type undivided cell (3 cm in diameter and 10 cm in height). A
constant current (20 mA, 2.83 F/mol) was passed for 140 min by using platinum foils (2 x 1.5 cm
2) as electrodes and a Metronix
Model 543B DC power supply. After the electrolysis at room temperature, 0.5 mL of 1% aqueous H
2SO
4 was added to the mixture, which was stirred for 1 h and neutralized with aqueous NaHCO
3. After evaporation of solvents under reduced pressure, the organic substances were extracted with ethyl acetate. The
extracts were washed with brine, dried (Na
2SO
4), and concentrated in vacuo to give a colorless oil, which was chromatographed (Mallinckrodt Silica CC-7 Special), affording
3a (116 mg, 98%) as colorless crystals
8 whose spectral data were identical with those reported. In an another experiment, after the electrolysis as mentioned above, the mixture was concentrated in vacuo and the organic substances were extracted with ethyl acetate. The usual
workup and chromatography provided
3a (28 mg, 23%) and
4 (78 mg, 71 %). The structure of
4 was identified spectroscopically by comparison with IR and
1H NMR spectra of the authentic sample prepared by mCPBA oxidation of
2.
92-Bromo-1-[3,4-(methylenedioxy)phenyl]-l-propanol (3b).A mixture of
2 (lOO mg) and NaBr (96 mg) dissolved in MeCN (7 mL)-1% aqueous H
2SO
4 (3 mL) was electrolyzed (20 mA for 125 min, 2.5 F/mol) in a similar manner as described above,
affording
3b9a (160 mg, quantitative yield) as a colorless oil: bp 110-112°C (0.02 mmHg); IR (neat) 3520, 3400 (OH), 1490, 1440, 1240, 1040, 790 cm
-1;
1H NMR (CDCl
3) ä 6.85 (br s, 1 H, Ar H), 6.77 (br s, 2 H, Ar H), 5.92 (s, 2 H, CH
2), 4.87 (d,
J = 4 Hz, 1 H, CHOH), 4.32 (dq,
J1 = 7 Hz,
J24 Hz, 1 H, CHBr), 2.54 (br s, 1 H, OH), 1.56 (d,
J = 7 Hz, 3 H, CH
3). Anal. Calcd for C
10H
11O
3Br: C, 46.35; H, 4.28. Found: C, 46.10; H, 4.20.
Electrolysis of 3a, Preparation of Piperonal (1).Glycol
3a (100 mg, 0.51 mmol) dissolved in benzene (6 mL) and 0.5% aqueous NaOH (4 mL) was electrolyzed at 65°C (18 mA for 3 h, 4 F/mol) in a similar manner as described above, affording
1 (76 mg, 99%) as colorless crystals whose spectral and TLC data
were consistent with those of an authentic sample.
Oxidation of 3a with NaIO4.A solution of NaIO
4 (82 mg, 0.38 mmol) was added to
3a (50 mg, 0.26 mmol) dissolved in MeOH (8 mL) at 0-5°C. The mixture was stirred at the temperature for 30 min. The usual workup provided
1 (36 mg, 94 % ).
Oxidation of 3a with CAN. Into a solution of
3a (100 mg, 0.51 mmol) dissolved in AcOH-H
2O (1:2,3 mL) was added CAN (587 mg, 1.07 mmol) dissolved in AcOH-H2O (1:2,12 mL). The mixture was stirred at room temperature for 30 min. The usual workup provided
1 (71 mg, 93% ).
Oxidation of 3a with Ca(ClO)2.A suspension of Ca(ClO)
2 (61 mg, 0.26 mmol) in benzene (2 mL)-H
2O (1 mL) was added to a solution of
3a (100 mg, 0.51 mmol) dissolved in benzene (4 mL)-H
2O (2 mL). After vigorous stirring at 65 °C for 1 h, Ca(ClO)
2 (183 mg, 0.77 mmol) suspended in benzene (2 mL)-H
2O (1 mL) was added again and the mixture was stirred for additional 2 h. After adding AcOEt and centrifuging a precipitate, the organic substances were extracted with AcOEt and the usual workup provided
1 (75 mg, 98%).
1-[2-Chloro-4,5-(methylenedioxy)phenyl]propane-l,2-diol (6).A suspension of Ca(ClO)
2 (60% purity, 92 mg, 0.39 mmol) in AcOH (0.12 mL)-H
2O (1.2 mL) was added drop wise to a solution of
3a (50 mg, 0.26 mmol) dissolved in MeCN (2 mL)-CH
2Cl
2 (1 mL). The reaction mixture was stirred at room temperature for 1 h. Ether extraction followed by usual workup and chromatography provided
6 (50 mg, 85%) as colorless crystals: mp 85-86°C: IR (CHCl
3) 3560 (OH), 3380 (OH), 1475, 1220, 1120, 1035, 935, 850 cm
-1;
1H NMR (CDCl
3) ä 6.96 (s, 1 H, Ar H), 6.80 (s, 1 H, Ar H), 5.97 (s, 2 H, CH
2), 4.86 (d,
J = 7 Hz, 1 H, CH), 3.84 (quint,
J = 7 Hz, 1 H, CH), 3.04 (br s, 1 H, OH), 2.64 (br s, 1 H, OH), 1.14 (d,
J = 7 Hz, 3 H, CH
3);
13C NMR (CDCl
3) ä 147.6 (s), 147.1 (s), 132.4 (s), 124.5 (s), 109.6 (d), 107.7 (d), 101.8 (t), 74.4 (d), 71.8 (d), 18.8 (q). Anal. Calcd for C
10H
11O
4Cl: C, 52.07; H, 4.81. Found: C, 52.26; H, 4.99.
1-[3,4-(Methylenedioxy)phenyl]-2-propanone (5). A solution of
3a (lOO mg, 0.5 mmol) and
p-TsOH (200 mg) dissolved in a distilled benzene (20 mL) was refluxed for 20 min. The usual workup gave
5 (75 mg, 84% ).
8,9Registry No. 1, 120-57-0;
2, 120-58-1;
3a, 62512-79-2;
3b, 57961-85-0;
5, 4676-39-5;
6, 89321-20-0; CAN, 16774-21-3; NaIO
4, 7790-28-5; Ca(ClO)
2, 7778-54-3.
References will be added during this week. From
https://www.thevespiary.org/rhodium/Rhodium/projects/typing/propenylbenzenes/isosafrole.electrochem.pdf

