Org. Lett., ASAP Article
DOI:
10.1021/ol036113f
Web Release Date: December 12, 2003
A General Synthesis of Substituted Indoles from Cyclic Enol Ethers and Enol Lactones Kevin R. Campos, Jacqueline C. S. Woo, Sandra Lee, and Richard D. Tillyer
Department of Process Research, Merck & Co., Inc., P.O. Box 2000, Rahway, New Jersey 07065 Received October 29, 2003 Abstract:
A general method was developed for the one-pot synthesis of highly functionalized indoles from simple, commercially available aryl hydrazines and cyclic enol ethers. Enol lactones were also used as substrates, affording substituted indole acetic acid or indole propionic acid derivatives. This procedure affords 2,3-disubstituted indoles as single regioisomers from the appropriately substituted enol ether or enol lactone. This method was highlighted in the efficient synthesis of the antimigraine drug sumitriptan and the antiinflammatory drug indomethacin.
The prevalence of indoles in natural products and biologically active compounds has led to a continued strong interest in the practical synthesis of the indole nucleus.
1 Among the diverse and creative approaches that have been discovered,
2 the Fischer indole reaction remains the benchmark to which other methods are compared.
3 Despite being quite versatile, the Fischer indole reaction with aldehydes often suffers from low yields and involves a two-step process (i.e., hydrazone formation, [3 + 3] rearrangement).
4 We herein wish to report a convenient and practical one-pot synthesis of 3-substituted indoles from commercially available cyclic enol ethers and enol lactones and the extension of this procedure to the regioselective synthesis of 2,3-disubstituted indoles (eq 1). This methodology is highlighted in the synthesis of several structurally diverse pharmaceutical agents, including the commercial drugs sumitriptan and indomethacin.
http://pubs.acs.org/isubscribe/journals/orlef7/asap/figures/ol036113ff1.html
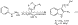
(
http://pubs.acs.org/isubscribe/journals/orlef7/asap/figures/ol036113ff1.html)
In our pursuit of an efficient synthesis of tryptophol homologs, we were intrigued with the possibility of using dihydropyran as an aldehyde equivalent in the Fischer indole reaction.
5 We suspected that suitable conditions could be developed that would not only generate the hydrazone from the aryl hydrazine and dihydropyran
in situ but also catalyze the [3 + 3] rearrangement in the same pot.
We chose 4% aqueous sulfuric acid as the solvent because of its documented success in promoting the Fisher indole reaction involving the
in situ hydrolysis of an aldehyde protected as its dimethyl acetal.
6 When dihydropyran was added to a solution of phenylhydrazine hydrochloride in 4% aqueous sulfuric acid at 100 °C, indole
2a was obtained in 50% isolated yield. The major byproduct of the reaction was triol
1, resulting from further reaction of
2a with dihydropyran (eq 2).
http://pubs.acs.org/isubscribe/journals/orlef7/asap/figures/ol036113ff2.html

(
http://pubs.acs.org/isubscribe/journals/orlef7/asap/figures/ol036113ff2.html)
We suspected that
1 was produced due to the high concentration of dihydropyran relative to indole product during the reaction; however, slow addition of dihydropyran did not decrease the level of
1 (49%).
7 After considerable study, it was discovered that addition of a cosolvent to the reaction significantly improved the reaction profile, producing less than 5% of byproduct
1.
8 Of the solvents investigated, acetonitrile (MeCN) and
N,
N-dimethylacetamide (DMAc) were optimal, affording indole
2a in 85 and 90% yields, respectively.
9The generality of the process was demonstrated with a wide variety of functionalized hydrazines bearing
ortho,
meta, and
para substituents (Table 1).
10 In the case of
m-tolyl hydrazine, a 1:1 mixture of regioisomeric indoles (
2c:
2d) was obtained. Aryl hydrazines bearing more than one substituent led to the formation of substituted indoles such as
2k in good yield.
The utility of this method was highlighted in the synthesis of Glaxo's antimigraine drug, sumitriptan (Scheme 1). Despite the documented difficulty of Fischer indole reactions with hydrazine
3 due to the instability of the product under acidic conditions,
11 the one-pot reaction could be accomplished to cleanly afford the desired hydroxyindole
4. Activation of the hydroxyl group as the mesylate followed by displacement in the presence of excess dimethylamine according to previously disclosed methodology
11b afforded sumitriptan in three steps and 45% overall yield (unoptimized) from dihydropyran.
Scheme 1. Synthesis of Sumitriptan
http://pubs.acs.org/isubscribe/journals/orlef7/asap/figures/ol036113fh00001.html

(
http://pubs.acs.org/isubscribe/journals/orlef7/asap/figures/ol036113fh00001.html)
To expand the scope of the methodology, the reaction of phenylhydrazine with other cyclic enol ethers was investigated (Table 2). For example, reaction of phenylhydrazine with dihydrofuran (
5) afforded tryptophol
6 in 72% yield. Interestingly, reaction of 1-methyldihydrofuran (
7) afforded the 2,3-disubstituted indole
8 in 83% isolated yield as a single regioisomer.
12Enol lactones also readily participate in this reaction. For example, reaction of enol lactone
11 with phenylhydrazine produced 2-methyl indole propionic acid
12 in 75% yield as a single regioisomer (Table 2, entry 4).
13 In contrast, when the procedure was attempted on angelicalactone (
13), the cyclic acyl hydrazone
14 was the only observed product.
14 This side reaction could be circumvented by installation of an
N-benzyl substituent on the hydrazine. Subjection of
N-benzyl phenylhydrazine to the same conditions cleanly afforded
N-benzyl-2-methylindole acetic acid (
16) in 70% yield.
This efficient approach to
N-protected indole acetic acids prompted us to apply the method to the synthesis of Merck's antiinflammatory drug, indomethacin.
15 Coupling of
N-acyl hydrazine
1716 with angelicalactone under standard conditions delivered indomethacin (
18) along with a significant amount of deacylated product. We discovered that reduction of the amount of water in the reaction suppressed the deacylation reaction. When the reaction was run with a minimal amount of water, using 1 equiv of sulfuric acid, indomethacin was produced in 65% yield (Scheme 2). This procedure represents a one-step approach to the regioselective synthesis of indomethacin from readily available starting materials.
Scheme 2. Synthesis of Indomethacin
http://pubs.acs.org/isubscribe/journals/orlef7/asap/figures/ol036113fh00002.html

(
http://pubs.acs.org/isubscribe/journals/orlef7/asap/figures/ol036113fh00002.html)
In conclusion, a general, one-pot method for the synthesis of highly functionalized indoles from commercially available cyclic enol ethers and enol lactones has been demonstrated. The procedure is general with respect to both the aryl hydrazine and the enol ether that are used. The method was especially useful for the regioselective synthesis of 2,3-disubstituted indoles. Cyclic enol lactones could be used in this coupling to afford substituted indole propionic acids and indole acetic acid derivatives in an efficient manner. The utility of this method was highlighted in the efficient synthesis of two commercial drugs: sumitriptan and indomethacin. We are currently investigating the application of this methodology to the preparation of highly functionalized indoles. The results of our findings will be reported in due course.
AcknowledgmentWe would like to thank Dr. Jeff Kuethe and Dr. Cheng-yi Chen (Merck & Co.) for valuable comments and discussions.
Detailed experimental procedures and compound characterization data. This material is available free of charge via the Internet at
http://pubs.acs.org/cgi-bin/suppinfo.pl?ol036113f
1. (a) Ihara, M.; Fukumoto, K.
Nat. Prod. Rep. 1995, 277. (b) Saxton, J. E.
Nat. Prod. Rep. 1994, 493. (c) Saxton, J. E.
Indoles; Wiley-Interscience: New York, 1983.
2. For a review of recent developments in the synthesis of indoles, see: Gribble, G. W.
J. Chem. Soc., Perkin Trans. 1.
2000, 1045-1075 and references therein.
3. (a) Gribble, G. W.
Contemp. Org. Synth. 1994,
1, 1. (b) Hughes, D. L.
Org. Prep. Proc. Int. 1993,
25, 607-632. (c) Robinson, B.
The Fischer Indole Synthesis; Wiley-Interscience: New York, 1982.
4. (a) Zepeda, L. G.; Morales-Rios, M. S.; Joseph,-Nathan, P.
Synth. Commun. 1992,
22, 3243-3256. (b) Shono, T.; Matsumura, Y.; Tsubata, K.
J. Org. Chem. 1984,
49, 3711-3716. (c) Fischer, G. W.
J. Heterocycl. Chem. 1995,
32, 1557-1561. (d) Pete, B.; Bitter, I.; Szantay, C. J.; Schon, I.; Toke, L.
Heterocycles 1998,
48, 1139-1149.
5. This method has been attempted, resulting in low yields as a two-step process. (a) White, J. D.; Yager, K. M.; Yakura, T.
J. Am. Chem. Soc. 1994,
116, 1831-1838. (b) McKittrick, B.; Failli, A.; Steffan, R. J.; Soll, R. M.; Hughes, R.; Schmid, J.; Asselin, A. A.; Shaw, C. C.; Noureldin, R.; Gavin, G.
J. Heterocycl. Chem. 1990,
27, 2151-2163.
6. (a) Chen, C.-Y.; Senanyake, C. H.; Bill, T. J.; Larsen, R. D.; Verhoeven, T. R.; Reider, P. J.
J. Org. Chem. 1994,
59, 3738-3741. (b) Klaver, W. H.; Hiemstra, H.; Speckamp, W. N.
J. Am. Chem. Soc. 1989,
111, 2588-2595.
7. Excess hydrazine (2 equiv) slightly reduced the amount of
1 (40%).
8. Importance of the cosolvent is due to the homogenization of the reaction mixture. In purely aqueous systems, the product and dihydrofuran are insoluble, creating a highly concentrated "organic layer" that leads to increased formation of
1.
9. Performing the reaction at 100 °C was optimal. Lower temperatures gave significantly lower yields of
2a (50 °C, 33% yield; 25 °C, 18% yield). However, in the case of more electron-rich hydrazines such as
p-methoxyphenylhydrazine, lower temperatures (55 °C) were needed to prevent the formation of the methoxy-substituted analogue of
1.
10.
Typical Procedure. To a solution of phenylhydrazine-HCl (1 g, 6.92 mmol) in 4% H
2SO
4 (aq) (10 mL) and DMAc (10 mL) at 100 °C was added dihydrofuran (630 uL, 6.92 mmol) dropwise over 2 min. The reaction was aged for 2 h and then cooled to room temperature, extracted with isopropyl acetate, and washed with water three times. The crude material was purified by flash chromatography.
11. Highest yield obtained in a Fischer indole with
3 was 63%. (a) Albinson, F. D.; MacKinnon, J. W. M.; Crookes, D. L. U.S. Patent 5103020, 1992. (b) Brodfuehrer, P. R.; Chen, B.-C.; Sattelberg, T. R.; Smith, P. R.; Reddy, J. P.; Stark, D. R.; Quinlan, S. L.; Reid, J. G.; Thottathil, J. K.; Wang, S.
J. Org. Chem. 1997,
62, 9192-9202.
[Full text - ACS]
(
http://pubs.acs.org/cgi-bin/citation?joceah/62/i26/html/jo971368q.html) Much lower yields (20-30%) have been reported in most other cases. (c) Dowles, M. D.; Coates, I. H. U.S. Patent 4816470, 1989. (d) Oxford, A. W. U.S. Patent 5037845.
12. Reported difficulty of achieving regioselective Fischer indole cyclizations on unsymmetrical ketones makes this result particularly noteworthy. Zhao, D.; Hughes, D. L.; Bender, D. R.; DeMarco, A. M.; Reider, P. J.
J. Org. Chem. 1991,
56, 3001 and references therein.
13. Structures containing this nucleus have been reported to be human neurokinin-1 receptor antagonists and serve as potential therapeutic agents for emesis, anxiety, and depression. Copper, L. C.; Chicchi, G. G.; Dinnell, K.; Elliott, J. M.; Hollingworth, G. J.; Kurtz, M. M.; Locker, K. L.; Morrison, D.; Shaw, D. E.; Tsao, K.-L.; Watt, A. P.; Williams, A. R.; Swain, C. J.
Bioorg. Med. Chem Lett. 2001, 1233-1236. Dinnell, K.; Chicchi, G. G.; Dhar, M. J.; Elliott, J. M.; Hollingworth, G. J.; Kurtz, M. M.; Ridgill, M. P.; Rycroft, W.; Tsao, K.-L.; Williams, A. R.; Swain, C. J.
Bioorg. Med. Chem. Lett. 2001, 1237-1240.
14. This is a commonly observed intermediate when the
N-acylhydrazone forms a six-membered heterocycle. Gouault, N.; Cupif, J. F.; Picard, S.; Lecat, A.; David, M.
J. Pharm. Pharmacol. 2001,
53, 981-985.
Medline (PMID=11480550)
15. Shen, T. Y.; et al
. J. Am. Chem. Soc. 1963,
85, 8-489. Shen, T. Y. U.S. Patent 3161654.
16. This hydrazine could be made in one step according to literature precedent in 90% yield from commercially available starting materials. Karady, S.; Ly, M. G.; Pines, S. H.; Chemerda, J. M.; Sletzinger, M.
Synthesis 1973, 50-51.
Table 1. Synthesis of 3-Substituted Indoles from Dihydropyran
http://pubs.acs.org/isubscribe/journals/orlef7/asap/figures/ol036113fu00001a.html

(
http://pubs.acs.org/isubscribe/journals/orlef7/asap/figures/ol036113fu00001a.html)
a All reactions were run in 4% H
2SO
4/DMAc (20 mL/g substrate) at 100 °C unless otherwise noted. All reported yields are after isolation by chromatography.
b Reaction was run in 4% H
2SO
4/MeCN (20 mL/g) at reflux.
c Reaction was run at 55 °C.
Table 2. Fischer Indole Reaction of Phenylhydrazine with Cyclic Enol Ethers and Enol Lactones
ahttp://pubs.acs.org/isubscribe/journals/orlef7/asap/figures/ol036113fu00002a.html

(
http://pubs.acs.org/isubscribe/journals/orlef7/asap/figures/ol036113fu00002a.html)
a All reactions were run in 4% H
2SO
4/DMAc (20 mL/g substrate) at 100 °C. All reported yields are after isolation by chromatography.
b N-benzyl phenylhydrazine was used in this coupling instead of phenylhydrazine.
c Reaction was run in 4% H
2SO
4/MeCN (20 mL/g substrate) at reflux.

