This post was edited to resolve stereoconfiguration confusion on September 3, 2002. /RhodiumAfter reading
J Chem Soc, 850-854 (1952), I found ideas for 4-Methylaminorex (4-MAR, U4EUh) syntheses, with or without the use of cyanogen bromide. All the examples below relates to the reaction of ephedrine and pseudoephedrine to form racemic 3,4-dimethylaminorex isomers, but the exactly same schemes should apply to phenylpropanolamine (PPA) to give 4-MAR. In this text, the designation phenylpropanolamine encompasses both norephedrine and norpseudoephedrine and their respective optical isomers. The two routes described are
1) The classic one-step cyanogen bromide cyclization of PPA and
2) Formation of the carbamyl (urea) derivative of PPA using potassium cyanate, followed by acid cyclization.
Using the cyanogen bromide route, norephedrine (1R,2S)/(1S,2R) yields cis-4-methylaminorex and norpseudoephedrine (1S,2S)/(1R,2R) yields trans-4-methylaminorex. The cyanogen bromide procedures below aren't optimized, addition of 3 equivalents of sodium acetate and doubling the amount of cyanogen bromide would not produce a precipitation of ephedrine salts, thus making the procedure more effective (see other syntheses of 4-MAR on my site). Also note that cyanogen bromide is very toxic.
Using the "double racemic" phenylpropanolamine (1RS,2RS) would give equal amounts of racemic cis- and trans-4-methylaminorex. All the cis/trans isomers are active, as well as their respective stereoisomers. The trans(4S,5S) is the most potent, with an effective dose of 0.25 mg/kg (compared to dextroamphetamine considered active at 0.4 mg/kg and d-meth at 0.2 mg/kg). The cis isomers are of about 5 times lesser potency, but they are still pretty active, as they are about equipotent to racemic amphetamine, and with a 2-3 times longer duration.
Using the potassium cyanate route, it should be noted that norephedrine (1R,2S)/(1S,2R) will be transformed into trans-4-MAR (the opposite of the cyanogen bromide route), while norpseudoephedrine (1S,2S)/(1R,2R) upon the same treatment instead forms trans-4-methyl-5-phenyl-oxazolid-2-one (which looks just like 4-MAR, but with a double bonded oxygen instead of the NH2 in the 2-position). It is an amide rather than an amine, so it should be removable using an acid-base extraction. Thus, no cis-4-MAR can be produced using potassium cyanate.
The byproduct amide trans-4-methyl-5-phenyl-oxazolid-2-one can be isolated and catalytically hydrogenated at room temperature in ethanol containing 5% triethylamine and 10 mol% Pd/C to form (S)-amphetamine in 90% yield, Ref:
Chem Eur J, 3( 1370 (1997)
1370 (1997), but that is probably not particularly useful, but using the potassium cyanate scheme on substituted (nor)ephedrines (like the 3,4-methylenedioxy variety) would enable you to produce a stereoselective synthesis of (S)-MDMA, the more active isomer, together with the 3,4-methylenedioxy-trans-4-MAR for evaluation of its activity.
Edit: The article can be found in Post 421261
(Rhodium: "Stereoselective (S)-MDA synth via the Cyanohydrin", Novel Discourse)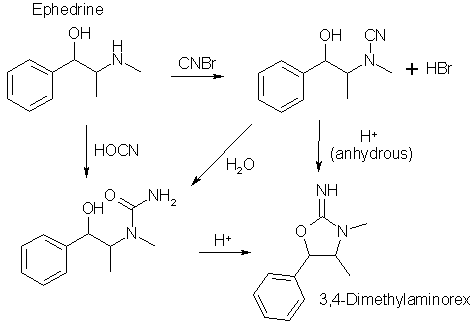 ExperimentalN-Carbamyl-(±)-ephedrine
ExperimentalN-Carbamyl-(±)-ephedrineTo 5g (±)-Ephedrine hydrochloride (25 mmol) in 25ml water was added 2g potassium cyanate (KOCN, 25 mmol), and the solution was heated under reflux for 2.5 hours, during which time a small amount of oil separated, then the solution was cooled in an ice-salt bath. The dried, white plates of the formed urea (3g, 57.7%) were recrystallized from ethyl acetate and was found to have a mp of 126-127°C.
(±)-trans-3,4-Dimethylaminorex HClA solution of N-carbamyl-(±)-ephedrine (1.56g, 7.5 mmol) in 24ml water and 15ml 2N HCl was refluxed for three hours, when the clear solution was cooled the (±)-trans-3,4-Dimethylaminorex hydrochloride precipitated. This was purified by basifying the solution, extracting it with benzene (can use any non-polar solvent here), and the solvent evaporated and the freebase converted to the hydrochloride by gassing with dry HCl in ether. Yield 1.9g, 84%, mp 225-229°C.
(±)-cis-3,4-Dimethylaminorex HCl (from (±)-ephedrine)60ml of an etheral solution containing 3.5g (30 mmol) cyanogen bromide was added to 200ml of an etheral solution containing 11g (±)-ephedrine (66 mmol), whereupon 8.1g of ephedrine hydrobromide separated (50% based on ephedrine input, 33 mmol, mp 186-188°C) and was filtered off and washed with ether. The filtrate was concentrated to 25ml, and white needles (1.5g, mp 71-73°C) of (±)-cis-3,4-Dimethylaminorex freebase precipitated. The filtrate was concentrated further, and the residual oil treated with ethanolic hydrogen chloride. The product was recrystallized from a mixture of 25ml CHCl3, 10 ml acetone and 5ml ether yielding 4.2g of (±)-cis-3,4-Dimethylaminorex hydrochloride, mp 215-217°C.
(±)-trans-3,4-Dimethylaminorex HCl (from (±)-pseudoephedrine)40 ml of an etheral solution of 1.75g cyanogen bromide (16.5 mmol) was added to a solution of 5.5g (±)-pseudoephedrine (33 mmol) in 100ml ether and 80ml benzene. In addition to precipitated pseudoephedrine hydrochloride (3.9g), (±)-cis-3,4-Dimethylaminorex hydrochloride (2.2g) was obtained upon treatment of the residual oil after evaporation of the solvent with ethanolic hydrogen chloride, mp 215-217°C, identical to the sample prepared above.

