SWIM has made some synthetic studies along the route proposed by
Kinetic (
Post 510530
(Kinetic: "Parham my mistake", Novel Discourse)).
1) 4-Hydroxybenzaldehyde was successfully alkylated. SWIM managed to get a 69% yield of a pure product using 1,2-dichloroethane in a non-optimized procedure. The published
1 procedure deals with 1-bromo-2-chloroethane, a more reactive alkylator, and only a 30% yield is achieved.
2) Bromination of the above aldehyde gave 40% of the monobromo product. The optimum reaction conditions are yet to be found.
3) The reaction of 3-bromo-4-(2-chloroethoxy)benzaldehyde with 4 eq. of EtMgBr in THF gave no cyclization product after a 2.5 h reflux, as can be inferred from the NMR spectra. Apparently, only addition of EtMgBr has occurred.
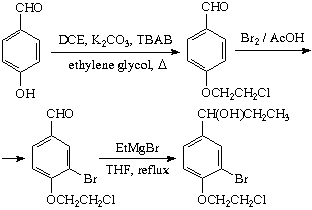
Of course, one can make a dimethyl acetal from 3-bromo-4-(2-chloroethoxy)benzaldehyde and cyclize it with Mg/THF as described above (
Post 510718
(Kinetic: "2,3-Dihydrobenzofurans without BuLi", Novel Discourse)).
Experimental part4-(2-Chloroethoxy)benzaldehydeBu
4NBr FW 322.36
1,2-Dichloroethane FW 98.96, d 1.256, bp 83°
4-Hydroxybenzaldehyde FW 122.12
K
2CO
3 FW 138.21
Na
2SO
3 FW 126.04
A 250 ml RBF equipped with a magnetic stirrer, a reflux condenser, and a gas bubbler connected to the top of the condenser was charged with 4-hydroxybenzaldehyde (18.06 g, 0.148 mol), anhydrous sodium sulfite (~0.2 g, 1.6 mmol), tetrabutylammonium bromide (2.39 g, 7.41 mmol, 5 mol. %), potassium carbonate (21.5 g, 0.156 mol), 1,2-dichloroethane (60 ml), and ethylene glycol (40 ml). The mixture was stirred at reflux for 24 h (at 17 h point gas evolution still continued, and TLC showed incomplete reaction). Then the mixture was cooled; water was added to dissolve the precipitated KCl, followed by toluene (60 ml). The contents of the flask were transferred to a separatory funnel, the flask was rinsed with toluene (10 ml), and the rinsings also were added to the separatory funnel. After shaking, the organic (upper) layer was separated and washed with 10% aq. KOH (4×25 ml). The aqueous layer was extracted with toluene (15 ml), and the extract was also washed with KOH solution (4×10 ml).
The combined organic phases were filtered
* through a mixture of silica gel (height 2 cm) and anhyd. Na
2SO
4 (height 0.5 cm) placed on a glass filter (diam. 4 cm); the adsorbed product was eluted with a mixture of toluene (80 ml) and ethyl acetate (20 ml), and the combined solutions were evaporated under reduced pressure to give a crude product (27.7 g) as a yellow oil, which solidified on standing in a refrigerator.
TLC (Merck F
254 SiO
2 plates, visualisation in UV light and with 0.5% 2,4-dinitrophenylhydrazine soln. in dil. H
2SO
4; eluent : CHCl
3 - Me
2CO 19:1 v/v) showed 2 major spots: R
f 0.65 (4-(2-chloroethoxy)benzaldehyde) and R
f 0.53 (presumably (OHCC
6H
4OCH
2)
2, since it was not detected in the product upon distillation), along with traces of 4-hydroxybenzaldehyde, R
f 0.24, and another aldehyde byproduct, R
f 0.19.
Distillation in a vacuum of an oil pump gave two fractions : bp 100-108 °C (1.83 g) and bp 108-115 °C (lit. bp 110 °C (0.1 mm Hg)
1; 138-142 °C (2 mm Hg)
2) . These were combined and redistilled. After a small forerun (0.96 g), the product was collected (
18.83 g, 69%; lit.
1 yield 30% from 1-bromo-2-chloroethane), which solidified on standing (mp 29-30 °C; lit.
1 mp 31°C). The smell is similar to that of anisaldehyde, but not so intense. TLC showed a small admixture of 4-hydroxybenzaldehyde. The forerun solidified below 20 °C.
1H NMR (200 MHz, CDCl
3): ? (ppm) 9.85 (s, 1H, CHO), 7.80 (m (AA'BB'), 2H, H-2, H-6), 6.98 (m (AA'BB'), 2H, H-3, H-5), 4.27 (t, 2H, J = 5.7 Hz, -OC
H2CH
2Cl), 3.82 (t, 2H, J = 5.7 Hz, -OCH
2C
H2Cl).
13C NMR (50 MHz, CDCl
3): ? (ppm) 190.56 (CHO), 162.94, 131.80, 130.21, 114.67 (benzene ring), 67.97 (-
CH
2O-), 41.48 (-
CH
2Cl).
*This step (actually, a short-column chromatography) was originally designed to get rid of Bu
4NBr and the byproduct having lower R
f value, presumably 4-(2-hydroxyethoxy)benzaldehyde. Instead, the solution can be simply dried with Na
2SO
4.
3-Bromo-4-(2-chloroethoxy)benzaldehyde.Br
2 FW 159.82, d 3.119
4-(2-Chloroethoxy)benzaldehyde FW 184.62, d
25 1.2246
2ZnCl
2 FW 136.28
A solution of bromine (1.8 ml, 35 mmol) in glacial AcOH (5 ml) was added to a solution of 4-(2-chloroethoxy)benzaldehyde (~5 ml, 6.18 g, 33.5 mmol) and zinc chloride (0.91 g, 6.7 mmol) in glacial AcOH (15 ml) dropwise with magnetic stirring over the course of 8 min (slight exothermy). The reaction flask was protected from light with aluminum foil. The mixture was allowed to stand at rt for 2 h 15 min. The product has virtually the same R
f value as the starting aldehyde in a number of eluents tested (CHCl
3 - Me
2CO 19:1, petroleum ether - EtOAc 7:3, benzene - EtOAc 9:1 v/v), so TLC appeared to be useless.
Another portion of Br
2 (0.4 ml, 7.8 mmol) was added (obviously, this was a mistake, see below), and the mixture was allowed to stand for 1.5 h. Then 2% aq. Na
2SO
3 was added with stirring until the bromine color disappeared; the volume of the mixture was brought to ~100 ml with water, the organic layer separated, and the aqueous layer extracted with CHCl
3 (3×10 ml).
The combined organic phases were washed with water (60 ml), 5% aq. NaOH containing 0.5% Na
2SO
3 (2×60 ml), dried (Na
2SO
4), and evaporated to give a colorless oil (7.75 g), which solidified on standing in a refrigerator. This was dissolved in a mixture of petroleum ether (20 ml), CCl
4 (10 ml) and PhMe (7 ml) at ~50 °C. On cooling to rt, crystals formed. The mixture was cooled to +4 °C; petroleum ether (10 ml) was gradually added to complete crystallization; the crystals were filtered off, washed with cold CCl
4, then with a 1 : 3 mixture of CCl
4 with petroleum ether, and dried.
Yield 3.585 g (40%) of white crystals, mp 84-85 °C, with a "pesticide" smell. R
f 0.37 (petr. ether - EtOAc 7:3 v/v).
The mother liquors and washings were evaporated to dryness. Low-temperature crystallization of the residue from methanol gave 1.95 g of white crystals with mp 50-53 °C, probably a dibrominated product, with R
f 0.57 (petr. ether - EtOAc 7:3 v/v). The melting point was raised to 53-54 °C after recrystallization from CCl
4 - petroleum ether. The solubility of the byproduct in CCl
4 is much higher than that of the main product. After standing at rt for several days, the crystals turned to a yellow liquid.
1H NMR (200 MHz, CDCl
3): ? (ppm) 9.84 (s, 1H, CHO), 8.06 (d, 1H, J = 1.9 Hz, H-2), 7.79 (dd, 1 H, J = 1.9 Hz, J = 8.5 Hz, H-6), 6.98 (d, 1H, J = 8.5 Hz, H-5), 4.36 (t, 2H, J = 5.9 Hz, -OC
H2CH
2Cl), 3.89 ((t, 2H, J = 5.9 Hz, -OCH
2C
H2Cl).
13C NMR (50 MHz, CDCl
3): ? (ppm) 189.43 (CHO), 159.21, 134.64, 131.08, 130.92, 113.01, 112.55 (benzene ring), 69.14 (-
CH
2O-), 41.11 (-
CH
2Cl).
1-(3-Bromo-4-(2-chloroethoxy)phenyl)-1-propanol3-Bromo-4-(2-chloroethoxy)benzaldehyde FW 263.52
EtBr FW 108.97, d 1.460
Mg FW 24.31
To a solution of EtMgBr prepared from EtBr (4.0 ml, 54 mmol) and Mg (1.93 g, 79 mmol) in abs. THF (50 ml) under argon was added a solution of 3-bromo-4-(2-chloroethoxy)benzaldehyde (3.48 g, 13.2 mmol) in abs. THF (20 ml) dropwise with stirring and cooling in a water bath (30-35 °C, internal temperature). The addition took 13 min. No precipitate has formed. The mixture was refluxed for 2.5 h (after 0.5 h TLC showed a single spot of the product, R
f 0.46 in CHCl
3 - Me
2CO 19 : 1 v/v; no changes were noted on further heating) and cooled to 0 °C. A saturated aq. solution of NH
4Cl (50 ml) was carefully added to the reaction mixture (Caution: ethane evolution!), followed by PhMe (50 ml). The organic layer was separated; the aqueous layer was extracted with PhMe (2×20 ml), and the combined extracts were washed with 10% aq. NaOH (3× 25 ml), dried (Na
2SO
4) and evaporated. The resulting product (viscous yellowish oil) gave a positive Beilstein test, and its NMR spectra were consistent with the title structure (
cf. 3 and
4).
1H NMR (200 MHz, CDCl
3): ? (ppm) 7.50 (s, 1H, H-2), 7.17 (d, 1H, J = 8.6 Hz, H-6), 6.84 (d, 1H, J = 8.6 Hz, H-5), 4.46 (t, 1H, J = 6.4 Hz, C
HOH), 4.25 (t, 2H, J = 6.0 Hz, -OC
H2CH
2Cl), 3.83 (t, 2H, J = 6.0 Hz, -OCH
2C
H2Cl), 2.54 (br. s, 1H, OH), 1.69 (m, 2H, -CHOH-C
H2CH
3), 0.86 (t, 3H, J = 7.3 Hz, CH
3).
13C NMR (50 MHz, CDCl
3): ? (ppm) 153.7, 139.2, 131.0, 127.2, 126.0, 114.4, 113.7, 112.3 (arom. ring; note the signals of admixtures), 74.6 (CHOH), 69.3 (-
CH
2O-), 41.5 (-
CH
2Cl), 31.7 (-CHOH
CH
2CH
3), 9.9 (CH
3).
1 J. Org. Chem.,
18, 1380 (1953).
2 Patent US2568579
.
3 The
13C NMR spectrum of 1-phenyl-1-propanol, found in
NMRShiftDB - NMR web database
(
http://www.nmrshiftdb.org/portal/pane0/Search), is as follows: 140.50, 126.53, 128.33, 127.76 (arom. ring), 77.30 (CHOH), 29.24 (CH
2), 9.84 (CH
3).
4 In the patent application
http://www.bandwidthmarket.com/resources/patents/apps/2001/7/20010006619.html
,
some
13C spectra of dihydrobenzofurans are presented. Thus, in 5-bromo-2,3-dihydrobenzo[
b]furan-7-carboxylic acid the chemical shift of C-2 is 71.78 ppm; that of C-3 is 27.88 ppm.
Nichols didn't publish the 13C NMR spectra of the benzofurans obtained by his group. In 1H spectra the chemical shifts of the benzylic CH2 groups of dihydrobenzofurans were at ~3.2 ppm.

