The "one-pot" reaction of (-)-phenylglycinol with formaldehyde in the presence of KCN gave the chiral 1,3-oxazolidine
(R)-
1 as an oil in 94% yield. This synthon was then alkylated twice in the ?-position of the cyano group using LDA/MeI followed by LDA/PhCH
2Br, the first substitution giving a product with 38%
d.e.* and the second >66%.
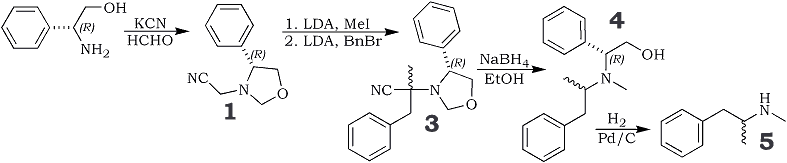
Reduction of the crude product
3 effected decyanation and ring-opening of the oxazolidine to give
4, which in essence is an N-alkylated methamphetamine molecule. If an enantiomerically pure end product is desired, a diastereomer separation is now performed to remove the 15% or so of
(R,R)-
4 from the remaining 85% of
(R,S)-
4 through flash chromatography (silica gel, eluting with 5% MeOH in DCM). The unwanted N-alkyl group is then finally removed through overnight hydrogenolysis at atmospherical pressure (MeOH, 10% Pd/C, 12h) to give
(S)-N-methyl-?-methyl-phenethylamine
5 (
d-Methamphetamine) in 90% yield, with preserved chirality.
The overall yield of
d-Methamphetamine from the starting
R-(-)-phenylglycinol is thus ~20%.
*d.e. = Diastereomeric Excess - A measure of diastereomer ratio, defined similarly to
enantiomeric excess.
(
http://www.wiu.edu/users/mftkv/Chem331/enantiomericexcess.html)
Asymmetric Synthesis IV.
Preparation of Chiral ?-Aminonitriles from a new N-Cyanomethyl-1,3-Oxazolidine Synthon
José L. Marco, Jacques Royer and Henri-Philippe Husson
Tetrahedron Letters 26(30), 3567-3570 (1985)
(https://www.thevespiary.org/rhodium/Rhodium/pdf/cyanomethyl-4-phenyl-13-oxazolidine.pdf)
Abstract: The synthesis of (-)-N-cyanomethyl-4-phenyl-1,3-oxazolidine 1 is reported. Good yields and moderate diastereomeric excesses of mono- and di-substituted ?-aminonitriles were obtained from this simple chiral template.
Preparation of (-)-N-cyanomethyl-4-phenyl-1,3-oxazolidine 1
To a stirred solution of (-)-phenylglycinol (23.62g, 0.16 mol), KCN (10.4g, 0.16 mol) in water (650 mL) at pH ~3 (citric acid) was added over 30 min at room temp a solution of formaldehyde (40%, 260 mL). The reaction mixture was stirred for ar additionnal 30 min, then basified (Na2CO3) and extracted (CH2Cl2). The combined organic fractions were washed with water, dried (Na2SO4) and concentrated to give a yellow oil which was purified by flash chromatography (SiO2, hexane-AcOEt, 80:20). 1 was obtained as a colorless oil (31.15g, 94% yield, [?]20D -173°(CHCl3, c=1.4)).
Alkylation of 1 with Methyl Iodide
To a stirred solution of LDA/HMPA (1:1, 1.1 eq. 0.48 M in THF) at -78°C, was added 1 (1 eq., 0.66M in THF) via syringe over 5 min; after 15 min 1.1 eq. of MeI was added. The reaction mixture was stirred for 1 h, quenched by NH4Cl then extracted with ether, dried and concentrated to dryness. Flash chromatography of the residual oil (SiO2, hexane-EtOAc, 85:15) separated the isomers to give 2 as the major product, yield 55% (38% d.e.) [?]20D -282° (c=2.4, CDCl3)
Asymmetric Synthesis IX:
Preparation of Chiral ?-Substituted Phenethylamines
J.L. Marco, J. Royer & H.-P. Husson
Synth. Commun. 17(6), 669-676 (1987)
(https://www.thevespiary.org/rhodium/Rhodium/chemistry/meth.n-cyanomethyloxazolidine.html)
Abstract: (S)-N-methyl-?-methyl-phenethylamines 5a-d were obtained in 56-62% e.e. from the chiral synthon (-)-N-cyanomethyl-4-phenyl-1,3-oxazolidine-1.
Alkylation of 2 with Benzyl Bromide
To a stirred solution of LDA/HMPA (1:1; 5 mmol, prepared from 3.12 mL of BuLi 1.6 M and 0.75 mL of diisopropylamine at -10°C/-30°C under a nitrogen atmosphere) in THF (20 mL) was added 2 (0.909g, 4.5 mmol, THF, -78°C) via syringe over 5 min. After 20 min, the resultant anion solution was added benzyl bromide (2 equiv.) and the mixture was stirred at -78°C for 1 h, quenched by NH4Cl then extracted with ether dried and concentrated to dryness. Flash chromatography of the residual oil (SiO2, hexane-AcOEt, 80:20) yielded compound 3, which was used in the next step without further purification.
Reductive Decyanation/Ring-Opening of 3
NaBH4 (1.1 eq.) was added portionwise to a solution of 3 in EtOH (~0.4 M); stirring was continued at room temp overnight. The solvent was removed and the residue taken up in DCM, washed with H2O and brine, dried over Na2SO4 and evaporated to dryness. The oily residue was purified by flash-chromatography (DCM-MeOH, 95:5) to give 4. The diastereomeric excess of the RS isomer was 66% and the overall yield from 1 was 30%.
Hydrogenolysis of 4 to give (S)-5 (d-Methamphetamine)
A methanolic solution (~0.2 M) of 4 was hydrogenolysed overnight at room temp, using 1 atm. H2 in the presence of 10% Pd/C. The reaction mixture was then filtered through a celite bed and the filtrate evaporated in vacuo to yield an oily residue which was purified by flash chromatography on silica (DCM-MeOH) to give (S)-5 (d-Methamphetamine) in 90% yield (66% e.e.).

