Here is the reference for the reduction of a secondary thioamide with Zn/HCl in EtOH (yield: 91%).
http://www.unibas.ch/mdpi/ecsoc-3/a0020/a0020.html
More on this topic will follow when Lego's alter ego will not bee busy during the opening times of the library.
N-METHYLTYRAMINE AND N,O-DIMETHYLTYRAMINE SYNTHESIS VIA INTERMEDIATE 4-METHOXYPHENYL-N-METHYLTHIOACETAMIDEVladimir N. Bulavka*, Alexander N. Shchavlinskii and Oleg N. Tolkachev
All-Russian Institute of Medicinal and Aromatic Plants (VILAR);
Grina str., 7, Moscow, 113628, Russian Federation.
*Present address: Scientific-Research Phototechnical Institute on Slavich Company (NIFTI-Slavich);
Mendeleev sq. 2, Pereslavl-Zalesskii, Yaroslavl region, 152140, Russian Federation.
E-mail:
nifti@slavich.botik.ru
Received: 30 July 1999 / Uploaded: 13 August 1999Keywords: galanthamine synthesis half-products, N-methyltyramine, N-methyltyramine ethers, N,O-dimethyltyramine, arylthioacetamide, Willgerodt-Kindler reaction
N-Methyltyramine and its alkyl or benzyl ethers are common intermediate products in the syntheses of narwedine, and galanthamine alkaloids [1-8]. They are usually produced from the corresponding arylacetamides by lithium aluminium hydride reduction. In order to avoid application of inflammable lithium aluminium hydride bulgarian scientists have transformed arylacetamide to arylthioacetamide with phosphorus (V) sulfide with following reduction of arylthioacetamide with complex sodium borohydride - nickel or cobalt chloride agents [7, 8].
Here we present a new short way route to N-methyltyramine and its ethers via the corresponding aryl-N-methylthioacetamides, shown at the scheme 1 at the example of N-methyltyramine and N,O-dimethyltyramine synthesis.
Scheme 1.
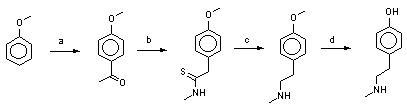
--> a: 92%
--> b: 57-58%
--> c: 91%
--> d: 98%
total 46-47% total 48-49%
a) H
3CCOBr, AlCl
3, ClCH
2CH
2Cl.
b) H
3CNH
2.HCl, S
8 , H
3CCO
2Na, (H
3C)
2NCHO, 115
oC, 3h.
c) Zn (dust) and 36% HCl(aq.) in ethanol
d) 36% HCl(aq.), refluxion
Anisole was smoothly acetylated in a classical conditions [9] with acetyl bromide and aluminium chloride in dichloroethane to produce 4-methoxyacetophenone in 92% yield.
Phenylacetic acid thioamides are usually synthesized by the Willgerodt-Kindler reaction from the corresponding substituted acetophenones, amines, and sulfur. We have studied several alternative routes to the 4-methoxyphenyl-N-methylthioacetamide synthesis shown at the scheme 2.
Scheme 2.
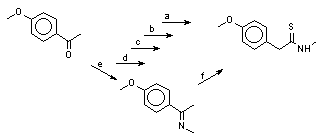
a) H
3CNH
2, S
8, 170-180
oC, 3h (yield 10,4%, and 21,9% by-product amide).
b) H
3CNH
2.HCl, S
8 , H
3CCO
2Na, (H
3C)
2NCHO, 115
oC, 3h (yield 58,4%).
c) H
3CNH
2.HCl, S
8, N(C
2H
5)
3, 4-H
3CC
6H
4SO
3H
.H
2O, (H
3C)
2NCHO, 75
oC, 72h (yield 57%)
d) H
3CNHCHO, S
8, 190
oC, 20h (yield 35%).
e) H
3CNH
2 .HCl, Na
2CO
3, CaO, N(C
2H
5)
3, -7
oC (isolated mixture imine : ketone 77:23 (
1H NMR) formed, used for the next stage without separation).
f) S
8, N(C
2H
5)
3, (H
3C)
2NCHO, 70
oC (total yield on two stages 21,5%).
The best results were produced according to routes b and c.
The key thioamide was isolated chromatographically on alumina or silica gel.
In classical conditions of the Willgerodt-Kindler reaction in the sealed tube [10] the desired thioamide was obtained in only 10.4% yield, while the corresponding by-product amide was also isolated in 21.9% yield. In order to avoid the application of high pressure, two-stage synthesis was carried out via intermediate 4-methoxyacetophenone methylimine [11] by method [12, 13] in 21.5% total yield.
Earlier proposed method of carrying out the Willgerodt-Kindler reaction with volatile lower secondary amines, e. g. dimethylamine, using its hydrochloride and sodium acetate in dimethyl formamide at 100-110
oC and atmospheric pressure was developed [14].
We have used a similar method for lower primary amine. Thus methylamine hydrochloride, 4-methoxyacetophenone, sulfur, and a base in dimethyl formamide at 60-110
oC and atmospheric pressure gave the desired thioamide in 57-58% yield.
An other route of 4-methoxyphenyl-N-methylthioacetamide synthesis on heating the mixture of 4-methoxyacetophenone, N-methylformamide and sulfur at 170-180
oC at atmospheric pressure according to the method [15] 35% yield was achieved.
4-Methoxyphenyl-N-methylthioacetamide is a new compound, forming colorless crystals, m. p. 89-91
oC (from ethanol or from diethyl ether) [16].
We have elaborated for the thioamide obtained a new simple and efficient method of reduction with zinc dust and hydrochloric acid in ethanolic solution. N,O-Dimethyltyramine was obtained in 91% yield, which was converted to its hydrochloride, m.p. 179-181
oC (from ethanol), (lit. m.p. 181-182
oC [17]).
O-Demethylation of N,O-dimethyltyramine to N-methyltyramine was carried out according to earlier described method for N-acetylated compound [18] by refluxing with concentrated hydrochloric acid in the oil bath heated to 170-175
oC. The yield was 98%, m. p. 146-149
oC (from ethanol with a

