Peracid Epoxidation of p-Methoxy-trans-?-Methylstyrene (trans-Anethole)Rebecca S. Centko and Ram S. MohanJ. Chem. Educ. 78(1), 77-78 (2001)Epoxidation of alkenes using peroxyacids is one of the most fundamental reactions in organic chemistry, yet there are very few examples of laboratory experiments that illustrate this important reaction (
Ref 1). The inherent instability of many epoxides in acidic solutions makes the synthesis of acid-sensitive epoxides by this route difficult. Frequently, the carboxylic acid formed from the peracid during epoxidation reacts with acid-sensitive epoxides to give ?-hydroxyesters as the major product. Procedures have been developed for epoxidation of alkenes in the presence of buffers to minimize this problem (
Ref 2).
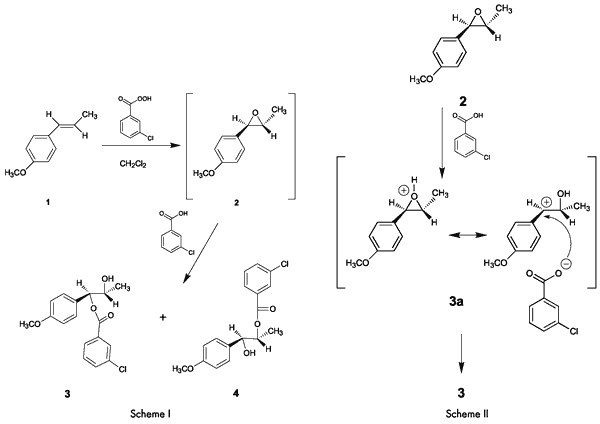
Overview of the ExperimentThe entire reaction, including the work up, takes only about an hour. Analysis of NMR and IR spectra of the product obtained in the absence of a buffer indicates that the ester, which has been assigned structure
3, is the major product.
The formation of
3 as the major product (
Note 1) rather than
4, can be attributed to a significant contribution to the resonance hybrid by the highly stable p-methoxy-substituted benzylic cation resonance form
3a (
Scheme II). This is consistent with the fact that in acid-catalyzed reactions, epoxides suffer attack at the carbon that can best stabilize positive charge (
Scheme II).
 Experimental SectionProcedure A (No Buffer)
Experimental SectionProcedure A (No Buffer)A solution of
trans-anethole (0.50 g, 3.4 mmol) in CH
2Cl
2 (10 mL) was stirred and cooled in an ice bath as a solution of MCPBA (0.92 g, 3.7 mmol) in CH
2Cl
2 (10 mL) was added dropwise. The resulting mixture was stirred in the ice bath for an additional 20 min. The mixture was washed with 10% Na
2CO
3 (5 x 15 mL) and saturated NaCl solution (15 mL). (
Note 2) The organic layer was dried (Na
2SO
4) and the solvent was removed on a rotary evaporator (
Note 2) to give 1.02 g (94%) of a viscous oil.
Procedure B (Buffered Epoxidation)A biphasic mixture of a solution of
trans-anethole (0.50 g, 3.4 mmol) in CH
2Cl
2 (10 mL) and 10% aqueous Na
2CO
3 solution (20 mL) was stirred well and cooled in an ice bath as a solution of MCPBA (1.4 g, 5.7 mmol, 1.7 equiv) in CH
2Cl
2 (20 mL) was added dropwise. After the addition was complete, the mixture was stirred for an additional 20 min in the ice bath. The organic layer was separated and washed with 10% aqueous Na
2CO
3 solution (5 x 25 mL) and saturated NaCl solution (15 mL). The organic layer was dried (Na
2SO
4) and the solvent was removed on a rotary evaporator to yield 0.52 g (95%) of a pleasant-smelling oil.
Notes1. Based on the 1H NMR spectrum, there appears to be only one major product, which has been assigned structure
3 on mechanistic grounds. The ester carbonyl can also be clearly seen in the IR spectrum of
3.
2. The excess peracid is removed by washing with 10% aqueous Na
2CO
3. The absence of peracid can be tested using starch-iodide paper.
3. Solvent can also be removed using a water bath maintained at 50°C.
The IR spectrum of the unbuffered reaction product shows the presence of an OH group and also an ester carbonyl, suggesting the formation of a hydroxy ester. What is the theoretical yield of the product, assuming it is the epoxide? How does this compare to the observed yields? The observed yield of product in the absence of buffer is almost twice the theoretical yield. This gives the first hint that the expected product has not formed. Rather, the higher mass recovery must be due to formation of a product with a much larger formula weight. Buffered epoxidation gives product in a yield comparable to the theoretical yield of the expected epoxide.
Further Notes* Commercial MCPBA, available from ACROS chemicals, is 70 % per acid by weight. We confirmed this by iodometric titration according to the procedure of Vogel (
Textbook of Practical Organic Chemistry, 5th Ed.; Vogel, A. I.; Longman Scientific and Technical: New York, 1989, p 455)
* Because
trans-anethole is very inexpensive, it was chosen rather than the much more expensive p-methoxystyrene.
* Peroxy acids (pKa ~8) are much weaker acids than carboxylic acids (pKa ~4). This allows for selective extraction of 3-chlorobenzoic acid. However, some of the MCPBA does react with Na
2CO
3, as is evident by the fact that the reaction does not go to completion when only 1 equiv. of MCPBA is used in presence of Na
2CO
3.
* The reaction mixture must be stirred very efficiently with a magnetic stir bar.
* If the organic layer is not extracted several times with Na
2CO
3, some of the 3-chlorobenzoic acid still remains in the organic layer.
* An alternative to the experiment described can be further purification of the ester by column chromatography (R
f 0.38, 40% ethyl acetate-60 % hexanes). The epoxide is not stable to silica gel.
* The ester and epoxide are not very volatile. Hence solvent can be easily removed using a water bath maintained at 50°C. The epoxide is very reactive and undergoes decomposition at higher temperatures.
Literature Cited1. For examples of epoxidation see Bradley, L. M.; Springer, J. W.; Delate, G. M.; Goodman, A.
J. Chem. Educ. 1997, 74, 1336. Garin, D. L.; Gamber, M.; Rowe, B.
J. Chem. Educ. 1996, 73, 555.
2. Mohan, R. S.; Whalen, D. L.
J. Org. Chem. 1993, 58, 2663-2669. Svensson, A.; Ulf, L. M.; Somfai, P.
Synth. Commun. 1996, 26, 2875–2880.

