2. Basicity of Amines
A review of basic acid-base concepts should be helpful to the following discussion. Like ammonia, most amines are Brønsted and Lewis bases, but their base strength can be changed enormously by substituents. It is common to compare basicities quantitatively by using the pKa's of their conjugate acids rather than their pKb's. Since pKa + pKb = 14, the higher the pKa the stronger the base, in contrast to the usual inverse relationship of pKa with acidity. Most simple alkyl amines have pKa's in the range 9.5 to 11.0, and their water solutions are basic (have a pH of 11 to 12, depending on concentration). The first four compounds in the following table, including ammonia, fall into that category.
The last five compounds (colored cells) are significantly weaker bases as a consequence of three factors. The first of these is the hybridization of the nitrogen. In pyridine the nitrogen is sp2 hybridized, and in nitriles (last entry) an sp hybrid nitrogen is part of the triple bond. In each of these compounds (shaded red) the non-bonding electron pair is localized on the nitrogen atom, but increasing s-character brings it closer to the nitrogen nucleus, reducing its tendency to bond to a proton.
Compound:
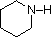
pK
a: 11.0
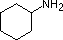
pK
a: 10.7
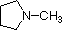
pK
a: 10.7
NH
3pK
a: 9.3

pK
a: 5.3
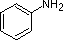
pK
a: 4.6

pK
a: 1.0
pK
a: 0.0
pK
a: -1.0

pK
a: -10.0
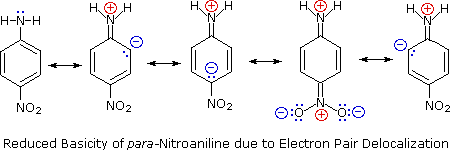
Secondly, aniline and p-nitroaniline (first two green shaded structures) are weaker bases due to delocalization of the nitrogen non-bonding electron pair into the aromatic ring (and the nitro substituent). This is the same delocalization that results in activation of a benzene ring toward electrophilic substitution. The following resonance equations, which are similar to those used to explain the enhanced acidity of ortho and para-nitrophenols illustrate electron pair delocalization in p-nitroaniline. Indeed, aniline is a weaker base than cyclohexyl amine by roughly a million fold, the same factor by which phenol is a stronger acid than cyclohexanol. This electron pair delocalization is accompanied by a degree of rehybridization of the amino nitrogen atom, but the electron pair delocalization is probably the major factor in the reduced basicity of these compounds. A similar electron pair delocalization is responsible for the very low basicity (and nucleophilic reactivity) of amide nitrogen atoms (last green shaded structure). This feature was instrumental in moderating the influence of amine substituents on aromatic ring substitution.
Finally, the very low basicity of pyrrole (shaded blue) reflects the exceptional delocalization of the nitrogen electron pair associated with its incorporation in an aromatic ring. Indole (pKa = -2) and imidazole (pKa = 7.0) also have similar heterocyclic aromatic rings. Imidazole is over a million times more basic than pyrrole because the sp2 nitrogen that is part of one double bond is structurally similar to pyridine, and has a comparable basicity.
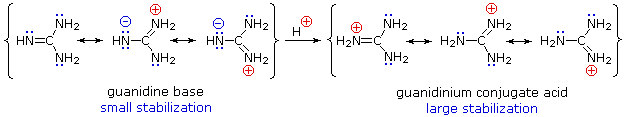
Although resonance delocalization generally reduces the basicity of amines, a dramatic example of the reverse effect is found in the compound guanidine (pKa = 13.6). Here, as shown below, resonance stabilzation of the base is small, due to charge separation, while the conjugate acid is stabilized strongly by charge delocalization. Consequently, aqueous solutions of guanidine are nearly as basic as are solutions of sodium hydroxide.
A similar delocalization of nitrogen and oxygen electron pairs by an adjacent carbonyl group was described in the context of substituent effects in aromatic substitution reactions. This interaction decreases the basicity of carbonyl derivatives of amines, called amides (pKa = -1), and will be discussed further in the section devoted to carboxylic acid derivatives.
3. Acidity of Amines
We normally think of amines as bases, but it must be remembered that 1º and 2º-amines are also very weak acids (ammonia has a pKa = 34). In this respect it should be noted that pKa is being used as a measure of the acidity of the amine itself rather than its conjugate acid, as in the previous section. For ammonia this is expressed by the following hypothetical equation:
NH
3 + H
2O ____> NH
2(–) + H
2O-H(+)
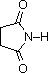
The same factors that decreased the basicity of amines increase their acidity. This is illustrated by the following examples, which are shown in order of increasing acidity. It should be noted that the first four examples have the same order and degree of increased acidity as they exhibited decreased basicity in the previous table. The first compound is a typical 2º-amine, and the three next to it are characterized by varying degrees of nitrogen electron pair delocalization. The last two compounds (shaded blue) show the influence of adjacent sulfonyl and carbonyl groups on N-H acidity. From previous discussion it should be clear that the basicity of these nitrogens is correspondingly reduced.
Compound:
pK
a: 33
pK
a: 27
pK
a: 19
pK
a: 15
C
6H
5SO
2NH
2pK
a: 10
pK
a: 9.6
The acids shown here may be converted to their conjugate bases by reaction with bases derived from weaker acids (stronger bases). Three examples of such reactions are shown below, with the acidic hydrogen colored red in each case. For complete conversion to the conjugate base, as shown, a reagent base roughly a million times stronger is required.
C
6H
5SO
2NH
2 + KOH C
6H
5SO
2NH(–) K(+) + H
2O a sulfonamide base
(CH
3)
3COH + NaH (CH
3)
3CO(–) Na(+) + H
2 an alkoxide base
(C
2H
5)
2NH + C
4H
9Li (C
2H
5)
2N(–) Li(+) + C
4H
10 an amide base
4. Important Reagent Bases
The significance of all these acid-base relationships to practical organic chemistry lies in the need for organic bases of varying strength, as reagents tailored to the requirements of specific reactions. The common base sodium hydroxide is not soluble in many organic solvents, and is therefore not widely used as a reagent in organic reactions. Most base reagents are alkoxide salts, amines or amide salts. Since alcohols are much stronger acids than amines, their conjugate bases are weaker than amide bases, and fill the gap in base strength between amines and amide salts.
Pyridine is commonly used as an acid scavenger in reactions that produce mineral acid co-products. Its basicity and nucleophilicity may be modified by steric hindrance, as in the case of 2,6-dimethylpyridine (pKa=6.7), or resonance stabilization, as in the case of 4-dimethylaminopyridine (pKa=9.7). Hünig's base is relatively non-nucleophilic (due to steric hindrance), and like DBU is often used as the base in E2 elimination reactions conducted in non-polar solvents. The alkoxides are stronger bases that are often used in the corresponding alcohol as solvent, or for greater reactivity in DMSO. Finally, the two amide bases see widespread use in generating enolate bases from carbonyl compounds and other weak carbon acids.
Amine Reactions
1. Electrophilic Substitution at Nitrogen
Ammonia and many amines are not only bases in the Brønsted sense, they are also nucleophiles that bond to and form products with a variety of electrophiles. A general equation for such electrophilic substitution of nitrogen is:
2 R2ÑH + E(+) R2NHE(+) R2ÑE + H(+) (bonded to a base)
A list of some electrophiles that are known to react with amines is shown here. In each case the electrophilic atom or site is colored red.
Electrophile
RCH2–X RCH2–OSO2R R2C=O R(C=O)X RSO2–Cl HO–N=O
Name
Alkyl Halide Alkyl Sulfonate Aldehyde or Ketone Acid Halide or Anhydride Sulfonyl Chloride Nitrous Acid
Alkylation
It is instructive to examine these nitrogen substitution reactions, using the common alkyl halide class of electrophiles. Thus, reaction of a primary alkyl bromide with a large excess of ammonia yields the corresponding 1º-amine, presumably by a SN2 mechanism. The hydrogen bromide produced in the reaction combines with some of the excess ammonia, giving ammonium bromide as a by-product. Water does not normally react with 1º-alkyl halides to give alcohols, so the enhanced nucleophilicity of nitrogen relative to oxygen is clearly demonstrated.
2 RCH2Br + NH3 (large excess) RCH2NH2 + NH4(+) Br(–)
It follows that simple amines should also be more nucleophilic than their alcohol or ether equivalents. If, for example, we wish to carry out a SN2 reaction of an alcohol with an alkyl halide to produce an ether (the Williamson synthesis), it is necessary to convert the weakly nucleophilic alcohol to its more nucleophilic conjugate base for the reaction to occur. In contrast, amines react with alkyl halides directly to give N-alkylated products. Since this reaction produces HBr as a co-product, hydrobromide salts of the alkylated amine or unreacted starting amine (in equilibrium) will also be formed.
2 RNH2 + C2H5Br RNHC2H5 + RNH3(+) Br(–) RNH2C2H5(+) Br(–) + RNH2
Unfortunately, the direct alkylation of 1º or 2º-amines to give a more substituted product does not proceed cleanly. If a 1:1 ratio of amine to alkyl halide is used, only 50% of the amine will react because the remaining amine will be tied up as an ammonium halide salt (remember that one equivalent of the strong acid HX is produced). If a 2:1 ratio of amine to alkylating agent is used, as in the above equation, the HX issue is solved, but another problem arises. Both the starting amine and the product amine are nucleophiles. Consequently, once the reaction has started, the product amine competes with the starting material in the later stages of alkylation, and some higher alkylated products are also formed. Even 3º-amines may be alkylated to form quaternary (4º) ammonium salts. When tetraalkyl ammonium salts are desired, as shown in the following example, Hünig's base may be used to scavange the HI produced in the three SN2 reactions. Steric hindrance prevents this 3º-amine (Hünig's base) from being methylated.
C6H5NH2 + 3 CH3I + Hünig's base C6H5N(CH3)3(+) I(–) + HI salt of Hünig's base
Reaction with Benzenesulfonyl chloride (The Hinsberg test)
Another electrophilic reagent, benzenesulfonyl chloride, reacts with amines in a fashion that provides a useful test for distinguishing primary, secondary and tertiary amines (the Hinsberg test). As shown in the following equations, 1º and 2º-amines react to give sulfonamide derivatives with loss of HCl, whereas 3º-amines do not give any isolable products other than the starting amine. In the latter case a quaternary "onium" salt may be formed as an intermediate, but this rapidly breaks down in water to liberate the original 3º-amine (lower right equation).
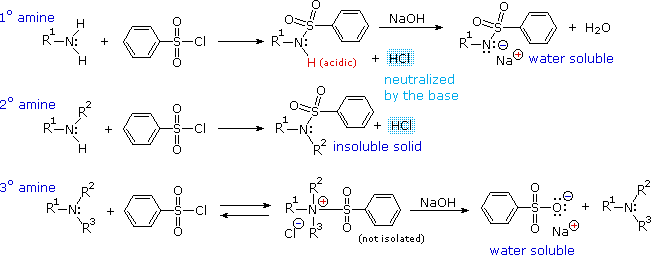
The Hinsberg test is conducted in aqueous base (NaOH or KOH), and the benzenesulfonyl chloride reagent is present as an insoluble oil. Because of the heterogeneous nature of this system, the rate at which the sulfonyl chloride reagent is hydrolyzed to its sulfonate salt in the absence of amines is relatively slow. The amine dissolves in the reagent phase, and immediately reacts (if it is 1º or 2º), with the resulting HCl being neutralized by the base. The sulfonamide derivative from 2º-amines is usually an insoluble solid. However, the sulfonamide derivative from 1º-amines is acidic and dissolves in the aqueous base. Acidification of this solution then precipitates the sulfonamide of the 1º-amine.
2. Preparation of 1º-Amines
Although direct alkylation of ammonia by alkyl halides leads to 1º-amines, alternative procedures are preferred in many cases. These methods require two steps, but they provide pure product, usually in good yield. The general strategy is to first form a carbon-nitrogen bond by reacting a nitrogen nucleophile with a carbon electrophile. The following table lists several general examples of this strategy in the rough order of decreasing nucleophilicity of the nitrogen reagent. In the second step, extraneous nitrogen substituents that may have facilitated this bonding are removed to give the amine product.
A specific example of each general class is provided in the diagram below. In the first two, an anionic nitrogen species undergoes a SN2 reaction with a modestly electrophilic alkyl halide reactant. For example #2 an acidic phthalimide derivative of ammonia has been substituted for the sulfonamide analog listed in the table. The principle is the same for the two cases, as will be noted later. Example #3 is similar in nature, but extends the carbon system by a methylene group (CH2). In all three of these methods 3º-alkyl halides cannot be used because the major reaction path is an E2 elimination.
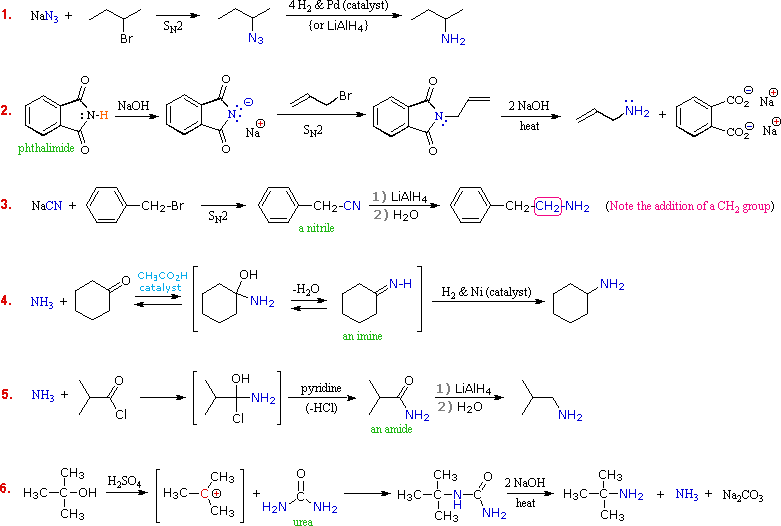
The methods illustrated by examples #4 and #5 proceed by attack of ammonia, or equivalent nitrogen nucleophiles, at the electrophilic carbon of a carbonyl group. A full discussion of carbonyl chemistry is presented later, but for present purposes it is sufficient to recognize that the C=O double bond is polarized so that the carbon atom is electrophilic. Nucleophile addition to aldehydes and ketones is often catalyzed by acids. Acid halides and anhydrides are even more electrophilic, and do not normally require catalysts to react with nucleophiles. The reaction of ammonia with aldehydes or ketones occurs by a reversible addition-elimination pathway to give imines (compounds having a C=N function). These intermediates are not usually isolated, but are reduced as they are formed (i.e. in situ). Acid chlorides react with ammonia to give amides, also by an addition-elimination path, and these are reduced to amines by LiAlH4.
The 6th example is a specialized procedure for bonding an amino group to a 3º-alkyl group (none of the previous methods accomplishes this). Since a carbocation is the electrophilic species, rather poorly nucleophilic nitrogen reactants can be used. Urea, the diamide of carbonic acid, fits this requirement nicely. The resulting 3º-alkyl-substituted urea is then hydrolyzed to give the amine.
One important method of preparing 1º-amines, especially aryl amines, uses a reverse strategy. Here a strongly electrophilic nitrogen species (NO2(+)) bonds to a nucleophilic carbon compound. This nitration reaction gives a nitro group that can be reduced to a 1º-amine by any of several reduction procedures.

