Well, I was going to keep this a secret until I could document it's success (if indeed it ever worked) and surprise the heck out of all you. But for a couple reasons it seems best to share the idea; most of all to get input from real chemists more knowledgeable, experienced, and probably smarter than I am  . (I also think it's likely that my reasoning is wrong, and if this train of thought isn't worth continuing, I'd prefer to know!)
. (I also think it's likely that my reasoning is wrong, and if this train of thought isn't worth continuing, I'd prefer to know!)
The idea is for a total synthesis of lysergic acid, and related compounds, which hinges upon a novel electrocyclization reaction. It is intended to use more common reagents than those of previous syntheses. The starting point is common to Rebek's 1983 synthesis: tryptophan, which is then hydrogenated to 2,3-dihydro-tryptophan, via Witkop's procedure with H2 and Pd/C, in order to protect the otherwise active 2-position. Similar hydrogenations with Raney Nickel were the starting points for earlier syntheses like Woodward's, leading me to believe a nickel-based catalyst may prove sufficient for this step. Dibenzoylation also protects the dihydrotryptophan, which is then cyclised to a ketone with Ac2O and AlCl3, according to the paper in 60% yield.
The branching point is the treatment of the ketone. Rebek proceeds with a Reformatsky reaction etc.. However, my idea is to couple it with an alkyne in preparation for formation of the D-ring; the reaction between ketones and terminal alkynes is well known and has a number of variations. A copper (I) based method may be used, however other possibilities for instance, some are based on zinc, others involve indium, and are of course to be used in different situations. My bet would tend to the copper, although trying to sort this out has been difficult and other possibilities may prove most efficient.
My original conception was to use propargyl alcohol or an ether, as the alkyne of choice, later to be oxidised to a carboxylic acid. But this would probably just make a mess. Lysergol has indeed been converted to lysergic acid by the gentlest means, in about one percent yield. It would probably be slightly better with the pyridine intermediate (I'll get to that), but at this point other ways seem better: using propiolic acid directly, or a protected version (perhaps the diethylamide straightaway), would make the most sense. The snag is, I'm not sure how this will modify its reaction with the ketone, or if it is in fact possible with propiolic acid – which would make propargyl alcohol/ether the best choice after all.
Pyrrole formation would have to be avoided, see "Syntheses and Reactions of Methyl Triphenylpyrrolecarboxylates" attached, which occurs (evidently) through Michael Condensation. So keep the sodium acetate away, haha.
The propargyl alcohol (where the ketone had been) would then be dehydrated to the alkene.
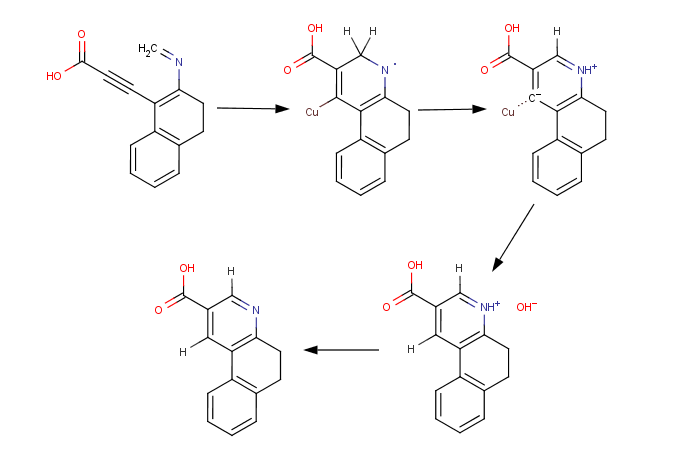
Then comes the real shaky bit, the cyclisation. The nitrogen would have to be deprotected, and if there's a way to selectively take off one but not the other, on the indoline-nitrogen, it might be better, but my assumption is that there isn't. In a nutshell, formaldehyde solution is used to form the imine, which cyclises under the influence of a copper-based catalyst. The tetracycle, presumably with an imine left on the indoline-nitrogen is then isolated and either hydrolysed back, or hydrogenated on it's way to form MLD-41. It can also be acetylated. But here, there are many variables and subtleties. The specificities of the catalyst, the temperature, and pH, are matters deserving focus and quite likely a large volume of experimentation, if it comes to that.
As to the pH, consider that at this point there are four main states for the molecule to be in; it may be positive, on the iminium ion, it can be negative (the carboxylic acid function deprotonated), it can be neutral, or the iminium and carboxylate functions are both present in the same molecule - a zwitterion or something, right?; each of which dramatically affects what happens when it reacts or doesn't at the catalytic site.
The idealised reaction mechanism is based on the neutral species; as the copper coordinates to the alkynyl carbons, the carbon closest to the carboxylic acid group draws electrons from the imine function, leaving the copper attached only to one. The temporary nitrogen radical is stabilised by interaction with another copper atom on the catalytic site, until an electron leaves the nearby methylene to complete the aromatic ring, which is negatively charged, and a postive hydrogen ion. Hydrolysis of the organocopper bond, which would seem to imply a relatively high temperature, replaces the unstable copper, which is now free to rest, with hydrogen, and a hydroxyl anion.
So many other things could happen, however, and if it at all worked it would be messy as shit. In the case of a carboxylate anion, I don't think anything important would go on, in the case of an iminium ion, I can imagine cyclisation but only to a diene. Etc. etc. I've spent rather some time thinking this bit through. The simplicity of the reagents involved would go some way toward countering this inefficience, and in any case, you can see how proper choice of pH and dilution would essential.
As for choice of catalyst, my instinct most strongly suggests copper on alumina, although I'm not sure why. However a variety of catalytic possibilities ought to be investigated, among them the possibility of a copper (I) substance, an halide or oxide possibly on a substrate or in solution, which would probably be somewhat more inclined to form the requisite intermediate anion complexes (CuCl2 1– is known for instance). Cu3BO3 also popped into my head, although I don't know if it exists, or if it does, in a form suitable for catalytic activity.
In any case, assuming it does anything, and the resulting “stuff” can be rectified into at least some amount of the pyridine substance (and in the case of the diene from more conventional electrocyclization of the iminium specie, would I be wrong in thinking it could be reduced into something more helpful?) the road ahead lies clear. The aromatic D-ring can be alkylated at the nitrogen, by methyl, ethyl, or what have you, and reduced by NaBH4 in acetonitrile as is done in Á Cobalt-Catalyzed Entry Into the Ergot Alkaloids, with 45% yield. In this case, the fact that there's an indoline ring, and that an amide function might not have been added, the yield might be much improved. Certainly it would be if the propargyl alcohol/ether route were used, and it would be as the pyridine substance that one would oxidise the alcohol to a carboxylic (nicotinic) acid, decreasing the chance of complications as I said.
Well that's the fundamental idea of it anyway. I would much appreciate feedback (pollination?). My version might be all impossible, but in that case I hope y'all could turn it into something viable. I'll include a couple pages that might be relevant to this. Rebek's synthesis is beautiful, and if you haven't already I recommend giving it a look.
On a side note, I wonder if the quaternary (TBAB) catalysed amide alkylation might be of any use in converting ergine and friends to the diethyl or other alkyl amides? The amine would definitely quaternise, but ethanolamine might be able to help. It sounds a little (rather) harsh but hell, you never know.
http://www.erowid.org/archive/rhodium/pdf/lsd.cobalt.pdf
http://www.organic-chemistry.org/synthesis/C1C/chains/propargylalcohols.shtm
http://www.organic-chemistry.org/synthesis/heterocycles/pyridines.shtm
http://books.google.com/books?id=WzhLRR-we-AC&lpg=PA51&ots=8fd1CoRPUz&dq=cyclisation%20of%20alkynes%20heterocycle&pg=PA78#v=onepage&q&f=true
Acetylene chemistry: chemistry, biology, and material science (Found on Google Books)
By François Diederich, Peter J. Stang, Rik R. Tykwinski
p.78&79 relevant to pyridines.
. (I also think it's likely that my reasoning is wrong, and if this train of thought isn't worth continuing, I'd prefer to know!)The idea is for a total synthesis of lysergic acid, and related compounds, which hinges upon a novel electrocyclization reaction. It is intended to use more common reagents than those of previous syntheses. The starting point is common to Rebek's 1983 synthesis: tryptophan, which is then hydrogenated to 2,3-dihydro-tryptophan, via Witkop's procedure with H2 and Pd/C, in order to protect the otherwise active 2-position. Similar hydrogenations with Raney Nickel were the starting points for earlier syntheses like Woodward's, leading me to believe a nickel-based catalyst may prove sufficient for this step. Dibenzoylation also protects the dihydrotryptophan, which is then cyclised to a ketone with Ac2O and AlCl3, according to the paper in 60% yield.
The branching point is the treatment of the ketone. Rebek proceeds with a Reformatsky reaction etc.. However, my idea is to couple it with an alkyne in preparation for formation of the D-ring; the reaction between ketones and terminal alkynes is well known and has a number of variations. A copper (I) based method may be used, however other possibilities for instance, some are based on zinc, others involve indium, and are of course to be used in different situations. My bet would tend to the copper, although trying to sort this out has been difficult and other possibilities may prove most efficient.
My original conception was to use propargyl alcohol or an ether, as the alkyne of choice, later to be oxidised to a carboxylic acid. But this would probably just make a mess. Lysergol has indeed been converted to lysergic acid by the gentlest means, in about one percent yield. It would probably be slightly better with the pyridine intermediate (I'll get to that), but at this point other ways seem better: using propiolic acid directly, or a protected version (perhaps the diethylamide straightaway), would make the most sense. The snag is, I'm not sure how this will modify its reaction with the ketone, or if it is in fact possible with propiolic acid – which would make propargyl alcohol/ether the best choice after all.
Pyrrole formation would have to be avoided, see "Syntheses and Reactions of Methyl Triphenylpyrrolecarboxylates" attached, which occurs (evidently) through Michael Condensation. So keep the sodium acetate away, haha.
The propargyl alcohol (where the ketone had been) would then be dehydrated to the alkene.
Then comes the real shaky bit, the cyclisation. The nitrogen would have to be deprotected, and if there's a way to selectively take off one but not the other, on the indoline-nitrogen, it might be better, but my assumption is that there isn't. In a nutshell, formaldehyde solution is used to form the imine, which cyclises under the influence of a copper-based catalyst. The tetracycle, presumably with an imine left on the indoline-nitrogen is then isolated and either hydrolysed back, or hydrogenated on it's way to form MLD-41. It can also be acetylated. But here, there are many variables and subtleties. The specificities of the catalyst, the temperature, and pH, are matters deserving focus and quite likely a large volume of experimentation, if it comes to that.
As to the pH, consider that at this point there are four main states for the molecule to be in; it may be positive, on the iminium ion, it can be negative (the carboxylic acid function deprotonated), it can be neutral, or the iminium and carboxylate functions are both present in the same molecule - a zwitterion or something, right?; each of which dramatically affects what happens when it reacts or doesn't at the catalytic site.
The idealised reaction mechanism is based on the neutral species; as the copper coordinates to the alkynyl carbons, the carbon closest to the carboxylic acid group draws electrons from the imine function, leaving the copper attached only to one. The temporary nitrogen radical is stabilised by interaction with another copper atom on the catalytic site, until an electron leaves the nearby methylene to complete the aromatic ring, which is negatively charged, and a postive hydrogen ion. Hydrolysis of the organocopper bond, which would seem to imply a relatively high temperature, replaces the unstable copper, which is now free to rest, with hydrogen, and a hydroxyl anion.
So many other things could happen, however, and if it at all worked it would be messy as shit. In the case of a carboxylate anion, I don't think anything important would go on, in the case of an iminium ion, I can imagine cyclisation but only to a diene. Etc. etc. I've spent rather some time thinking this bit through. The simplicity of the reagents involved would go some way toward countering this inefficience, and in any case, you can see how proper choice of pH and dilution would essential.
As for choice of catalyst, my instinct most strongly suggests copper on alumina, although I'm not sure why. However a variety of catalytic possibilities ought to be investigated, among them the possibility of a copper (I) substance, an halide or oxide possibly on a substrate or in solution, which would probably be somewhat more inclined to form the requisite intermediate anion complexes (CuCl2 1– is known for instance). Cu3BO3 also popped into my head, although I don't know if it exists, or if it does, in a form suitable for catalytic activity.
In any case, assuming it does anything, and the resulting “stuff” can be rectified into at least some amount of the pyridine substance (and in the case of the diene from more conventional electrocyclization of the iminium specie, would I be wrong in thinking it could be reduced into something more helpful?) the road ahead lies clear. The aromatic D-ring can be alkylated at the nitrogen, by methyl, ethyl, or what have you, and reduced by NaBH4 in acetonitrile as is done in Á Cobalt-Catalyzed Entry Into the Ergot Alkaloids, with 45% yield. In this case, the fact that there's an indoline ring, and that an amide function might not have been added, the yield might be much improved. Certainly it would be if the propargyl alcohol/ether route were used, and it would be as the pyridine substance that one would oxidise the alcohol to a carboxylic (nicotinic) acid, decreasing the chance of complications as I said.
Well that's the fundamental idea of it anyway. I would much appreciate feedback (pollination?). My version might be all impossible, but in that case I hope y'all could turn it into something viable. I'll include a couple pages that might be relevant to this. Rebek's synthesis is beautiful, and if you haven't already I recommend giving it a look.
On a side note, I wonder if the quaternary (TBAB) catalysed amide alkylation might be of any use in converting ergine and friends to the diethyl or other alkyl amides? The amine would definitely quaternise, but ethanolamine might be able to help. It sounds a little (rather) harsh but hell, you never know.
http://www.erowid.org/archive/rhodium/pdf/lsd.cobalt.pdf
http://www.organic-chemistry.org/synthesis/C1C/chains/propargylalcohols.shtm
http://www.organic-chemistry.org/synthesis/heterocycles/pyridines.shtm
http://books.google.com/books?id=WzhLRR-we-AC&lpg=PA51&ots=8fd1CoRPUz&dq=cyclisation%20of%20alkynes%20heterocycle&pg=PA78#v=onepage&q&f=true
Acetylene chemistry: chemistry, biology, and material science (Found on Google Books)
By François Diederich, Peter J. Stang, Rik R. Tykwinski
p.78&79 relevant to pyridines.