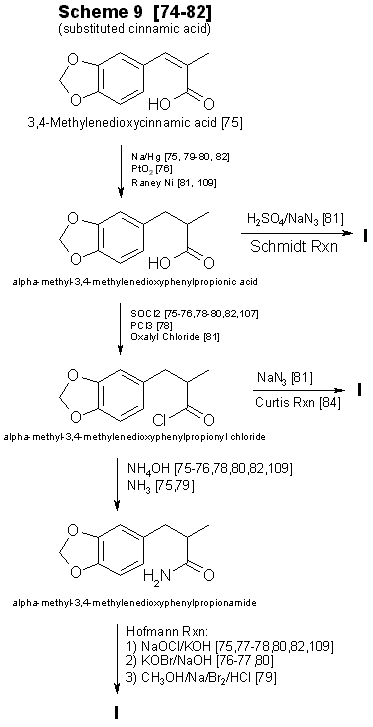
Ok, we have all had one or more encounters with trying to get that fucking methyl alcohol off vanillin (presumption). The difficulty is and will always be, that the vulnerable aldehyde is just that, vulnerable.
Now, I'm also presuming that most everyone here can also find (http://www.sciencemadness.org/talk/viewthread.php?action=printable&fid=10&tid=9706) Klute's sterling work on the condensation of Acetone & Vanillin to get the Zingerone, whereas if one used HCl/MEK one would presumably get the equivalent a-methyl-phenylbutanone. Now, peracid oxidations aren't high on my list of things to even contemplate with crap/no glass, but I'm bloody certain I've seen more than one paper where the hypochlorite oxidation of the methyl ketone would result in 3-methoxy-4-hydroxy-a-methylcinnamic acid. Now, there really shouldn't be any great difficulty in demethylating the a-methylcinnamic acid (if there is, reduction with boiling ethanol Ni/C should get rid of the double-bond).
That would give 3,4-dihydroxyphenyl-2-methyl-propanoic acid (or something like that - the a-methylcaffeic acid - http://en.wikipedia.org/wiki/Caffeic_acid - or the hydrocaffeic acid). Reduction (if the double bond is still there), then methylation with DCM/KI/etc. should give a bloody robust intermediate.
3,4-methylenedioxy-a-methylhydrocaffeic acid should form an amide easily enough, hypochlorite oxidation of that would be a bad idea as it would give MDA.
Attached there is an electrolytic procedure for reducing cinnamic acid in 90% yield, which should do. I'm waiting on a number of references, I know Woodruff & Conger used hypochlorite to reach a-methylcinnamic acid & they also made the amine by the Hoffman degradation of the hydrogenated amide. So that part is documented. I'll have a look around for a decent system for ether cleavage, but HI/HBr would do it (once the double-bond is gone). Simple, easy, no hard to get anything - or the need for massive amounts of equipment outlay.
Now, I'm also presuming that most everyone here can also find (http://www.sciencemadness.org/talk/viewthread.php?action=printable&fid=10&tid=9706) Klute's sterling work on the condensation of Acetone & Vanillin to get the Zingerone, whereas if one used HCl/MEK one would presumably get the equivalent a-methyl-phenylbutanone. Now, peracid oxidations aren't high on my list of things to even contemplate with crap/no glass, but I'm bloody certain I've seen more than one paper where the hypochlorite oxidation of the methyl ketone would result in 3-methoxy-4-hydroxy-a-methylcinnamic acid. Now, there really shouldn't be any great difficulty in demethylating the a-methylcinnamic acid (if there is, reduction with boiling ethanol Ni/C should get rid of the double-bond).
That would give 3,4-dihydroxyphenyl-2-methyl-propanoic acid (or something like that - the a-methylcaffeic acid - http://en.wikipedia.org/wiki/Caffeic_acid - or the hydrocaffeic acid). Reduction (if the double bond is still there), then methylation with DCM/KI/etc. should give a bloody robust intermediate.
3,4-methylenedioxy-a-methylhydrocaffeic acid should form an amide easily enough, hypochlorite oxidation of that would be a bad idea as it would give MDA.
Attached there is an electrolytic procedure for reducing cinnamic acid in 90% yield, which should do. I'm waiting on a number of references, I know Woodruff & Conger used hypochlorite to reach a-methylcinnamic acid & they also made the amine by the Hoffman degradation of the hydrogenated amide. So that part is documented. I'll have a look around for a decent system for ether cleavage, but HI/HBr would do it (once the double-bond is gone). Simple, easy, no hard to get anything - or the need for massive amounts of equipment outlay.

 If xxxxxxxx becomes scheduled the method in my last post should still be assessable though this I'd think. Anyways, after extraction I'd assume one could methylenate the caffeic acid like a catechol and subsequently hydrogenate the resulting product for 3,4-MD-phenylpropionic acid. I adapted a couple existing procedures (refs available) to hopefully result in the desired carboxylic acid...
If xxxxxxxx becomes scheduled the method in my last post should still be assessable though this I'd think. Anyways, after extraction I'd assume one could methylenate the caffeic acid like a catechol and subsequently hydrogenate the resulting product for 3,4-MD-phenylpropionic acid. I adapted a couple existing procedures (refs available) to hopefully result in the desired carboxylic acid...