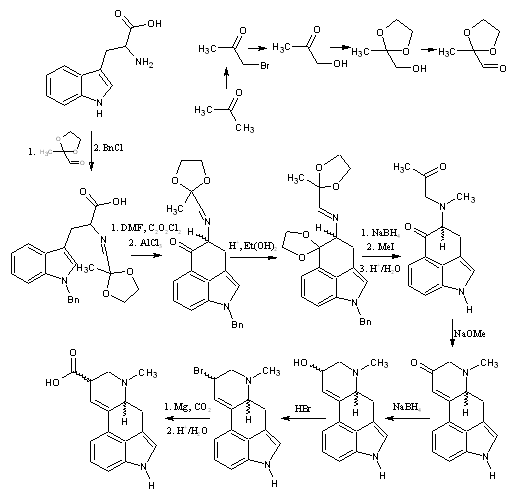
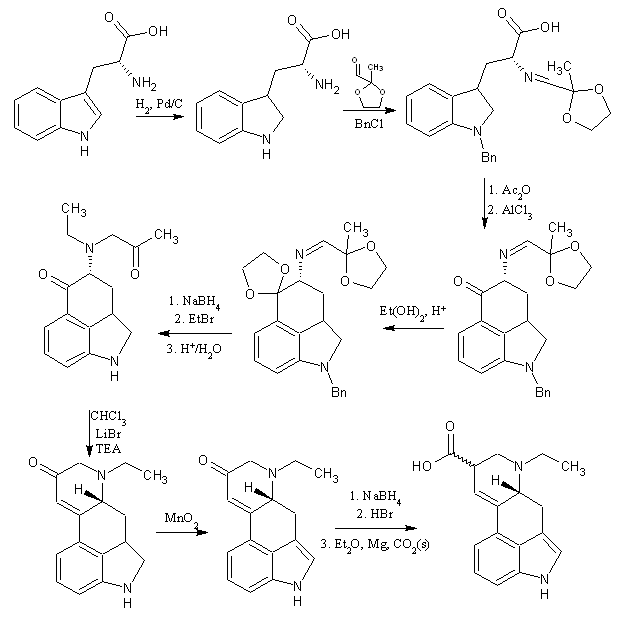
The drawing assumes that racemic tryptophan is used, which probably wouldn't happen. L-tryptophan is both OTC and produces the correct chirality on one of the chiral centers of the final product, so the yield is only halved once.
1/2 mole of tryptophan would be about 100g, and 500ml of acetone could be reacted into about 1/2 mole of the (not sure on name) ethylene-glycol protected acetone 3-aldehyde. If a 60-80% yield average could be maintained over the subsequent steps, it could be a 1-15% conversion of tryptophan to 1:1 lysergic acid:isolysergic acid.
Here are procedures from rhodium and orgsyn for the first two steps of of the acetone product:
After this, the protection of the carbonyl could probably be done using ethylene glycol as the solvent with some sulfuric acid. After refluxing for a while, distill off the ethylene glycol, perhaps under vacuum and extract with ether(?). This product would then be oxidized with something like IBX in DMSO. If the solvent used to extract this product was also compatible with tryptophan and had > water boiling point, the final extract could be maybe used more easily in the next step.
1/2 mole of tryptophan would be about 100g, and 500ml of acetone could be reacted into about 1/2 mole of the (not sure on name) ethylene-glycol protected acetone 3-aldehyde. If a 60-80% yield average could be maintained over the subsequent steps, it could be a 1-15% conversion of tryptophan to 1:1 lysergic acid:isolysergic acid.
Here are procedures from rhodium and orgsyn for the first two steps of of the acetone product:
Quote
A 5-L, three-necked, round-bottomed flask is provided with an efficient mechanical stirrer, a 48-cm. Allihn reflux condenser, a thermometer, and a 500ml separatory funnel, the stem of which reaches nearly to the bottom of the flask.
Through the separatory funnel are introduced 1.6 1. of water, 500ml of pure acetone, and 372 ml of glacial acetic acid. The stirrer is started and the temperature of the water bath is raised to 70-80°C, so that the mixture in the flask is at about 65°C. Then 354 ml (7.3 moles) of bromine is carefully added through the separatory funnel. The addition, which requires one to two hours, is so regulated as to prevent the accumulation of unreacted bromine As a rule the solution is decolorized in about twenty minutes after the bromine has been added. When the solution is decolorized, it is diluted with 800 ml of cold water, cooled to 10°C, made neutral to Congo red with about 1 kg. of solid anhydrous sodium carbonate, and the oil which separates is collected in a separatory funnel and dried with 80g of anhydrous calcium chloride. After drying, the oil is fractionated and the fraction boiling at 38-48°C/13 mmHg is collected. The yield is 470-480 g. (50-51% yield). If a purer product is desired, the above product is refractionated and the fraction boiling at 40-42°C/13 mmHg is collected. The yield is 400-410 g. (43-44% yield).
The higher-boiling fraction contains a mixture of isomeric dibromoacetones.
Quote
In a 3-l. round-bottomed flask fitted with a 75-cm. Liebig condenser is placed 210 g. of potassium hydroxide (purified with alcohol) dissolved in 1.5 l. of anhydrous methyl alcohol. The solution is cooled to below 50° (Note 1), 300 g. of purified ethyl formate is added, and the mixture is refluxed for two hours (Note 2) and (Note 3).
Then 410 g. (251 cc., 3 moles) of bromoacetone (p. 88) is added, and the mixture is refluxed for sixteen hours on a water bath at 95–97°. At the end of the operation the solution is cooled to 0° in an ice-salt bath. The potassium bromide which settles is filtered on a cooled suction filter, and the filtrate is fractionated.
The fraction boiling at 23–35°/12 mm. is discarded, as it contains very little acetol. The main fraction distils at 35–47°/12 mm. and weighs 160 g. This material is refractionated, and the portion boiling at 40–43°/12 mm. is collected. The yield is 120–130 g. (54–58 per cent of the theoretical amount) (Note 4).
After this, the protection of the carbonyl could probably be done using ethylene glycol as the solvent with some sulfuric acid. After refluxing for a while, distill off the ethylene glycol, perhaps under vacuum and extract with ether(?). This product would then be oxidized with something like IBX in DMSO. If the solvent used to extract this product was also compatible with tryptophan and had > water boiling point, the final extract could be maybe used more easily in the next step.