ImAMANGUYS: you've attempted this reaction before? did you just get unreacted safrole back out of it?
What was your workup technique?
Others have said Bicarb is not the best -- because it creates emulsions from hell.
I did get some emulsion last night but it broke fairly easily with brine and sitting still.
To be honest, I'm probably not the person you should be asking as i'm experimenting myself as well. But I'll share a small writeup I did with you if you want to compare results, then maybe someone with success at this method will chip in. I'm starting to become curious about how many members have had success with it. To answer your questions and for comparison here ya go.
BromoSafrole Attempt 1
Reactants:
(10ml, 0.067 mol) Safrole
(14.58g, 0.1417 mol) NaBr
(6.869ml, 12.64g, 0.1289mol) H2SO4
(24.82ml) GAA 40% w/w
Writeup:
To a 500ml flask was added 24.82ml GAA. To this, 14.58g NaBr was added and stirred vigorously. This mixture was cooled till the GAA began to freeze, at which point the 6.869ml H2SO4 was added drop wise. An orange hue was noted. The color was expected due to the off-clear sulfuric acid used, and the small amounts of Br2 formed. This solution:
Equation: H2SO4 + NaBr = NaHSO4 + HBr
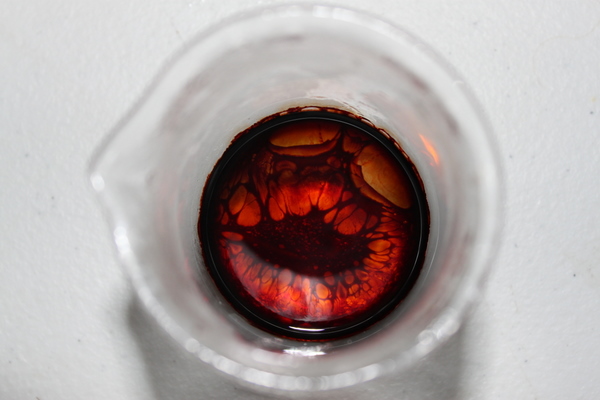
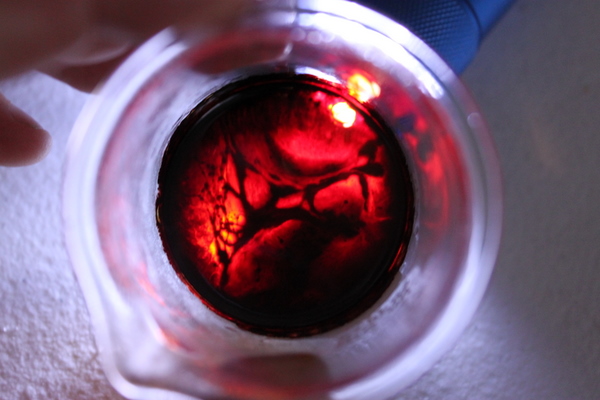
was mixed and left to cool for 30 minutes in a salt water ice bath. Temp approx 0*C. The flask was removed and the 10 ml of safrole was added all at once, a slightly green color (colors off in the pic) was noted.
(SEE PICTURE 1)
This mixture was left undisturbed in a salt water ice bath for the next 24 hours. The temp of the ice bath varied between -1C to 2C during this time, not exceeding or descending below these numbers. The flask was removed from the bath, at which point a blue-purple hue was observed. Because no indication has been described on the forums about how to tell the reaction has completed, and i've read about successful purple hues in the past, I assumed it was done. In honesty, I don't know how you can tell without decanting and testing on cold H2SO4. This after 24 hours.
(SEE PICTURE 2)
Workup was done quickly after pics were taken and the flask had been removed from the bath. The liquid was decanted from the salts:
(See PICTURE 3)
The first water was, quenching the mixture, resulted in a number of layers:
(SEE PICTURE 4)
Which upon standing and stirring, resulted in two layers:
(SEE PICTURE 5)
The mixture smelled of safrole, and the water layer remained milky.
I removed the bottom carmel colored layer and washed with sodium bicarbonate, at which point bubbling and fizzing did occur. Like directions indicate, I added till no fizzing was noted. I did another water wash, at which point the oil was yellowish brown.
When tested on Cold H2SO4, the mixture sat on top for a few a good 15 seconds before releasing tiny bits of purple. Maybe it just wasn't given enough time to react?
From the smell and color, it was a failed workup or reaction.
Hopefully someone with more experience can bring us both insight. Until then good luck my friend!
















 Embarrassing Stuff.
Embarrassing Stuff.