



Here is an idea. Please critique it.
134g (1 mol) 1-phenylpropan-2-one
150g (2 mol) glycine
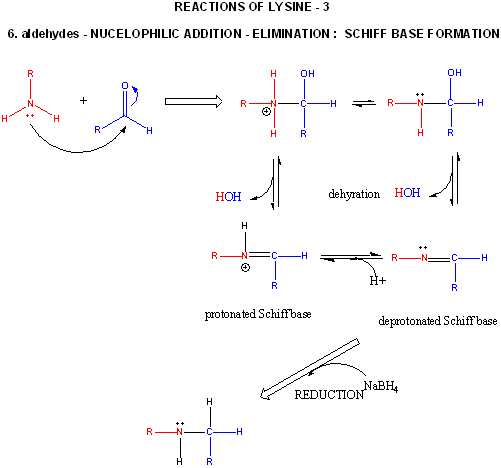
Mixed to form slurry. Flask flushed with nitrogen and gently brought to super slow reflux. Temperature held steady until reflux stops indicating complete formation of the imine. <6hrs time.
Reaction flask cooled in freezer. 100mL cold dry methanol added to post reaction mess, stirred and filtered. Add an additional 400mL cold dry methanol, place in ice with stirring and begin adding
15g (.4 mol, 160% needed hydride eq.) sodium borohydride
at a rate slow enough to keep reaction below 40C. Continue stirring for an hour after fizzing stops. Acidfy with HCl, filter, extract with ether, keeping aq. Base with NaOH and extract with DCM, keeping DCM. Brine, dry with magnesium sulfate and HCl gas for glory.
Reaction of p2p and glycine, from Bulletin de la Societe Chimique de France (1965), (4), 929-33,
“Similarly, an amino acid suspension in a high boiling ketone (nonanone, cyclo-hexanone, acetophenone, 2-, 3-, and 4-methyleyclohexanone, p-methylacetophenone, benzyl methyl ketone, propiophenone, benzophenone) yielded by decarboxylation, a Schiff base which was hydrolyzed in 3N HCl to regenerate the ketone and to give the amine-HCl.”
A reflux of p2p at atmospheric pressure should be hot enough to drive the produced water right out the top of the column, while refluxing p2p. The imine has a boiling point substantially higher, so as the reaction mix changes from p2p to imine, the gentle reflux and slight boiling should stop, sort of like watching the isomerazation of safrole.
This reaction mix will be thick and I’m sure will turn brown/black. As long as what we’re after is in there, no matter. Most reactions aren’t pretty.
That French ref was using refluxing mixtures of 50mL solvent to 5g amino acid, quite a bit more watered down than my proposed paste. I hesitate from using one of the high boiling point solvents only to make the post reaction separation easier. But I guess if the paste fails, a non ketone paraffin, decalin, tetralin or something with a high boiling point, but not so close to the bp of the imine as to be un separable by vac distillation. I think it would be tough to get them cleanly separated.
Filtration of post reaction mess by methanol should only pull the good stuff and leave glycine/carbonized mess behind. Merck index says that 100g absolute alcohol pulls .06g glycine. I don’t think imine will be helping the solubility of it any so that is less than .0008mol carboxylic acid to reduce to ethylamine with the sodium borohydride. Perhaps an additional rinse of the filter cake with cold dry methanol is needed.
Glycine is insoluble in ether, so another option would be extraction with ether and reduction with lithium aluminum hydride. Or ether extraction and vacuum evaporation.
On to the reduction, Hudlickey Reductions in Organic Chemistry p209 and the work of LabTop both use the 60% excess over the needed hydride equivalent. I propose using less than half the methanol used in LabTops write-ups mostly because there is no need to keep methylamine in solution in this case. I also don’t understand the need to keep this reaction below 20C as stated by LabTop. I have never read the refs he listed so I don’t know whether that temperature came from experience or reading.
One other question is whether the methanol imine mix should have a pellet or two of NaOH tossed in before the NaBH4 to help keep it from reacting with the methanol, even though from reading that is a reducing agent as well.
I read that wasps/bees are missing that mix the hive had, but remember the yin yang of that was the library know-it-all’s and legitimate working chemists who would never break a law or even do drugs sharing ideas and knowledge with those assholes willing and able to experiment and report.
Those assholes might be reading! So speak up, or don’t.
134g (1 mol) 1-phenylpropan-2-one
150g (2 mol) glycine
Mixed to form slurry. Flask flushed with nitrogen and gently brought to super slow reflux. Temperature held steady until reflux stops indicating complete formation of the imine. <6hrs time.
Reaction flask cooled in freezer. 100mL cold dry methanol added to post reaction mess, stirred and filtered. Add an additional 400mL cold dry methanol, place in ice with stirring and begin adding
15g (.4 mol, 160% needed hydride eq.) sodium borohydride
at a rate slow enough to keep reaction below 40C. Continue stirring for an hour after fizzing stops. Acidfy with HCl, filter, extract with ether, keeping aq. Base with NaOH and extract with DCM, keeping DCM. Brine, dry with magnesium sulfate and HCl gas for glory.
Reaction of p2p and glycine, from Bulletin de la Societe Chimique de France (1965), (4), 929-33,
“Similarly, an amino acid suspension in a high boiling ketone (nonanone, cyclo-hexanone, acetophenone, 2-, 3-, and 4-methyleyclohexanone, p-methylacetophenone, benzyl methyl ketone, propiophenone, benzophenone) yielded by decarboxylation, a Schiff base which was hydrolyzed in 3N HCl to regenerate the ketone and to give the amine-HCl.”
A reflux of p2p at atmospheric pressure should be hot enough to drive the produced water right out the top of the column, while refluxing p2p. The imine has a boiling point substantially higher, so as the reaction mix changes from p2p to imine, the gentle reflux and slight boiling should stop, sort of like watching the isomerazation of safrole.
This reaction mix will be thick and I’m sure will turn brown/black. As long as what we’re after is in there, no matter. Most reactions aren’t pretty.
That French ref was using refluxing mixtures of 50mL solvent to 5g amino acid, quite a bit more watered down than my proposed paste. I hesitate from using one of the high boiling point solvents only to make the post reaction separation easier. But I guess if the paste fails, a non ketone paraffin, decalin, tetralin or something with a high boiling point, but not so close to the bp of the imine as to be un separable by vac distillation. I think it would be tough to get them cleanly separated.
Filtration of post reaction mess by methanol should only pull the good stuff and leave glycine/carbonized mess behind. Merck index says that 100g absolute alcohol pulls .06g glycine. I don’t think imine will be helping the solubility of it any so that is less than .0008mol carboxylic acid to reduce to ethylamine with the sodium borohydride. Perhaps an additional rinse of the filter cake with cold dry methanol is needed.
Glycine is insoluble in ether, so another option would be extraction with ether and reduction with lithium aluminum hydride. Or ether extraction and vacuum evaporation.
On to the reduction, Hudlickey Reductions in Organic Chemistry p209 and the work of LabTop both use the 60% excess over the needed hydride equivalent. I propose using less than half the methanol used in LabTops write-ups mostly because there is no need to keep methylamine in solution in this case. I also don’t understand the need to keep this reaction below 20C as stated by LabTop. I have never read the refs he listed so I don’t know whether that temperature came from experience or reading.
One other question is whether the methanol imine mix should have a pellet or two of NaOH tossed in before the NaBH4 to help keep it from reacting with the methanol, even though from reading that is a reducing agent as well.
I read that wasps/bees are missing that mix the hive had, but remember the yin yang of that was the library know-it-all’s and legitimate working chemists who would never break a law or even do drugs sharing ideas and knowledge with those assholes willing and able to experiment and report.
Those assholes might be reading! So speak up, or don’t.
 .
.