The route to CH3CONH2 does proceed thru CH3COONH4 on at least one occasion. Various literature states that one method used in preparing acid amides or in this instance more specifically CH3CONH2 is by the dehydration of its corresponding ammonium salt. So imo it would be possible to utilize 1 of the 2 more well-known routes used when discussing routes to CH3CONH2. One thinks this is somewhat similar to Lugh's suggestion however in this process dehydration is accomplished within the synth itself (unless I am missing something???) either way you would need: 15g of Ammonium carbonate, 15 g.; acetic acid, and a 50 ml.of GAA.
Add 15 g. of finely powdered ammonium carbonate gradually to 50 ml. of glacial acetic acid contained in a 150 ml. rb flask, shaking the mixture during the addition to ensure a steady evolution of carbon dioxide. When all the carbonate has dissolved and the evolution of carbon dioxide has ceased, proceed to setup your flask for reflux by utilizing an appropriate open-air condenser, boil the solution gently for 3o minutes, when the dehydration of the ammonium acetate will be complete. Now remove the condenser and fit a fractionating column to the flask (at this point the literature which is a bit dated does make strong mention of the need for an efficient fractionating column. They proceed with providing dimensions for fabricating such devices, which imho probably dates this synth well before many of our equally effective modern day devices not only became more easily accessible but also helped establish standards, therefore an abbreviated version of this dialogue equates to an efficient column with the "height of about 12 cm. is used". Now distill the liquid through the column very slowly (to give about 1 drop of distillate every 3 seconds) until the temperature reaches 17o°: it is very important that this operation should be carried out slowly in order to distil over as much water and excess of acetic acid as possible, and so to leave almost pure acetamide in the flask. Without delay, pour the hot molten acetamide into a small (5o ml.) distilling-flask, and attach the sidearm of the flask to a short glass tube (about 3o cms. long and t2 mm. internal diameter) to act as an condenser. Distill the acetamide carefully at such a rate that none escapes condensation, and that none crystallises while still in the condenser. Collect the fraction boiling between 215° and 225° Yield, 15 g.
The later part of this synth. would obviously be omitted for your needs however should one not have a problem with either the availability of reagents or the requirement of a little manual dexterity by shaking a container followed by the ability to set-up a simple reflux apparatus, then hopefully this is what you are looking for. Hey I am still looking for one on ph strips so dont feel bad.
Not sure if the Urea route mentioned would be viable in acquiring CH3COONH4 however another type of ammonium does turn up in the Urea method and that is NH4NH2CO2 however since one has recently become a die-hard hofmann fan and loves the idea of synths starting from the floor up, then you could safely assume he would jump at the chance and say something like;
In regards to acetamide via urea, one could try placing 25 g. of glacial acetic acid and 25 g. of urea in a 100 ml. triple neck rb flask. Fit the center neck with an appropriate air-cooled condenser set for reflux. In either side-arms insert your thermometer so that the bulb in the mixture and 1 cm. from the bottom of the flask, close off the remaining neck with appropriate device such as a rubber septum. Heat the mixture gently either on a wire gauze or in an air bath. When the urea melts, shake the flask gently in order to mix the acid and urea layers. Gradually raise the temperature so that the liquid just refluxes in the condenser. The temperature is about 150° after 30 minutes and a whitesolid (probably ammonium carbamate) commences to form in the condenser : push the solid back into the flask by means of a stout glass rod when complete blocking of the condenser appears likely. Continue the heating until the temperature of the liquid is 195-200° ; this temperature is attained after a heating period of 3-3.5 hours. Both carbon dioxide and ammonia are evolved. Allow the apparatus to cool and rearrange it for distillation. Heat the flask slowly at first ; some ammonium carbamate first sublimes into the air condenser. When the acetamide begins to just reach the condenser, stop the distillation momentarily, replace the condenser by another of similar size and continue the distillation. Collect the acetamide at 200-216° (most of it passes over at 214-216°) ; if it crystallizes in the condenser, it may be melted by the cautious application of a flame. The yield of almost pure, colourless acetamide, m.p. 80.5°, is 22 g.
much love
mez






 You need to dry it under a vacuum, keeping the temperature low enough to avoid decomposition:
You need to dry it under a vacuum, keeping the temperature low enough to avoid decomposition: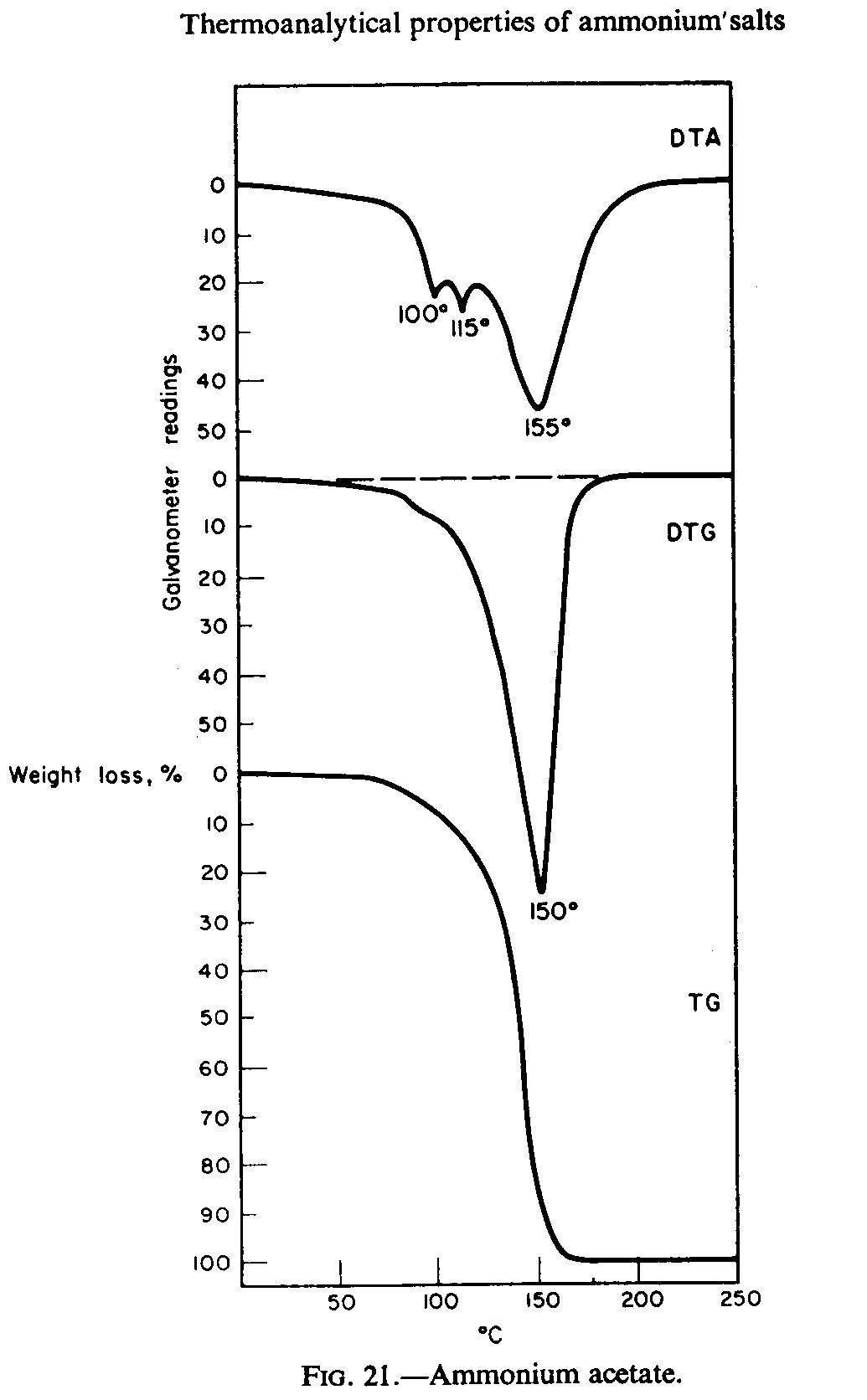
 and that aint going to fucking happen, no offence to collectors btw
and that aint going to fucking happen, no offence to collectors btw