



Decarboxylation of alpha Amino acids to their corresponding amines is not new, most of this research has been directed towards tryptophan but any a-Amino Acid can be used with slightly reduced yields. Decarboxylation of said compounds can be traced back to the sixties. Back in those days they refluxed the amino acid (tryptophan in this case) and diphenyl ether in a Nitrogen atmosphere to afford 57% yields of the amine.
J. Chem. Soc. 3990-94 (1965)
http://www.erowid.org/archive/rhodium/pdf/tryptophan2tryptamine.ph-o-ph.pdf
There are many other examples, for instance copper or zinc acetate with DMSO yields around 40%
http://www.erowid.org/archive/rhodium/chemistry/tryptamine.kametani-2.html
A newer route that deserves some attention is the novel decarboxylation of a-Amino acids in high yield, the reaction seems to be catalyzed by the presence of 2-cyclohexen-1-one created in tiny amounts from a small 3% peroxide addition to cyclohexanol then refluxing with a-Amino acid.

http://www.journalarchive.jst.go.jp/english/jnlabstract_en.php?cdjournal=cl1972&cdvol=15&noissue=6&startpage=893
Obviously this process could be used for alot of different purposes;
N,N-Dimethylation of the commonly available tryptophan with paraformaldehyde and oxalic acid, followed by the above treatment with cyclohexanol would afford pure DMT in high yields.
N-methyl-a-methyl-D-phenylalanine would produce (R)-methamphetamine in high yields! It would be easier to N-methylate after the fact, so alpha methyl-D-phenylalanine would produce dextroamphetamine which could then be methylated to enantiomerically pure meth in high yield. Traditional methods of substitutions to the alpha carbon of a-amino acids require amine protection, then oxazolidinone formation followed by enolization which leaves an electron rich double bond where we want it, for alkyl halides (methyl iodide in our case) are electrophilic (electron lovers) and will attach to the electron rich double bond, due to the new stability of the molecule the halide leaving group finds better options and leaves as a stable hydrohalogen. Acid hydrolysis opens the ring and the protecting group removed to leave a-methyl Phenylalanine. There are similar procedures that negate the need for a protecting group by using carbonyl chlorides to catalyze the oxazolidinone ring formation. But carbonyl chlorides, are generally insidious poison gasses such as Phosgene or Tri-Phosgene.
Synthesis of alpha-methyl, alpha-subtituted amino acids
Patent number; 6043376
Process for the preparation alpha-alkylated alpha-amino acids and alpha-halogenated amino acids
Patent number; 5153358
I have come across a number of other methylation procedures that proceed via the alpha carbon, leaving a-methyl amino acids. But so far require reagents or techniques that are out of the grasp of us for the time being. I will get back to you on that, also with any more easy to do substituted phenethylamines via this route I come across.
This also leaves open many routes to substituted phenethylamines, such as 4-HO-DMT (Psilocin) and many mescaline derivatives.
J. Chem. Soc. 3990-94 (1965)
http://www.erowid.org/archive/rhodium/pdf/tryptophan2tryptamine.ph-o-ph.pdf
There are many other examples, for instance copper or zinc acetate with DMSO yields around 40%
http://www.erowid.org/archive/rhodium/chemistry/tryptamine.kametani-2.html
A newer route that deserves some attention is the novel decarboxylation of a-Amino acids in high yield, the reaction seems to be catalyzed by the presence of 2-cyclohexen-1-one created in tiny amounts from a small 3% peroxide addition to cyclohexanol then refluxing with a-Amino acid.
http://www.journalarchive.jst.go.jp/english/jnlabstract_en.php?cdjournal=cl1972&cdvol=15&noissue=6&startpage=893
Obviously this process could be used for alot of different purposes;
N,N-Dimethylation of the commonly available tryptophan with paraformaldehyde and oxalic acid, followed by the above treatment with cyclohexanol would afford pure DMT in high yields.
N-methyl-a-methyl-D-phenylalanine would produce (R)-methamphetamine in high yields! It would be easier to N-methylate after the fact, so alpha methyl-D-phenylalanine would produce dextroamphetamine which could then be methylated to enantiomerically pure meth in high yield. Traditional methods of substitutions to the alpha carbon of a-amino acids require amine protection, then oxazolidinone formation followed by enolization which leaves an electron rich double bond where we want it, for alkyl halides (methyl iodide in our case) are electrophilic (electron lovers) and will attach to the electron rich double bond, due to the new stability of the molecule the halide leaving group finds better options and leaves as a stable hydrohalogen. Acid hydrolysis opens the ring and the protecting group removed to leave a-methyl Phenylalanine. There are similar procedures that negate the need for a protecting group by using carbonyl chlorides to catalyze the oxazolidinone ring formation. But carbonyl chlorides, are generally insidious poison gasses such as Phosgene or Tri-Phosgene.
Synthesis of alpha-methyl, alpha-subtituted amino acids
Patent number; 6043376
Process for the preparation alpha-alkylated alpha-amino acids and alpha-halogenated amino acids
Patent number; 5153358
I have come across a number of other methylation procedures that proceed via the alpha carbon, leaving a-methyl amino acids. But so far require reagents or techniques that are out of the grasp of us for the time being. I will get back to you on that, also with any more easy to do substituted phenethylamines via this route I come across.
This also leaves open many routes to substituted phenethylamines, such as 4-HO-DMT (Psilocin) and many mescaline derivatives.







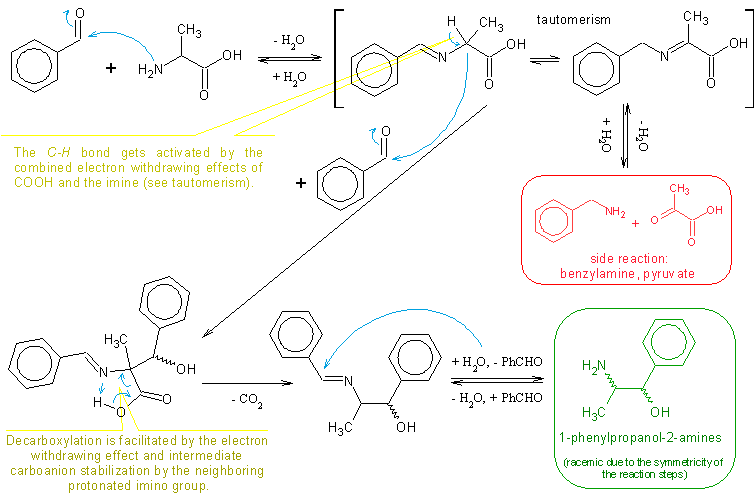


 but that only looks like)! Notice, that in both resonance structures the double bond does indicate the place where positive charge is delocalised, while the third atom(having its electron pair) is the one to be attacked with LA. Besides positive charge is carried by carbon also(carbocation), that is another important resonance structure not mentioned on the pic. So, the next lewis acid moiety would attach to either of nitrogen or oxygen, but not carbon.
but that only looks like)! Notice, that in both resonance structures the double bond does indicate the place where positive charge is delocalised, while the third atom(having its electron pair) is the one to be attacked with LA. Besides positive charge is carried by carbon also(carbocation), that is another important resonance structure not mentioned on the pic. So, the next lewis acid moiety would attach to either of nitrogen or oxygen, but not carbon. 

 It is nice that someone is interested in understanding the mechanisms of reactions. On most drug resources people like to cook and mechanisms are bit neglected. But i am always glad to explain them for those wasps who are interested.
It is nice that someone is interested in understanding the mechanisms of reactions. On most drug resources people like to cook and mechanisms are bit neglected. But i am always glad to explain them for those wasps who are interested.