phew.... everything went fine 
picked it up this morning, there wasnt even pressure on the bottle
picked it up this morning, there wasnt even pressure on the bottle
 Topic: Short Question Thread 2.0 (Read 5707 times)
Topic: Short Question Thread 2.0 (Read 5707 times)



I´M lost again.- Formation of the nitric acid ester followed by catalytic hydrogenation with hydrogen preloaded Pd on BaSO4. That should be the most practical hydrogenation pathway.
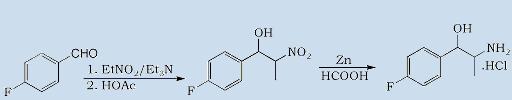
I had discovered recently the Nitroalcohol route to Phenylpropanolamine. (BzCOH +EtNO2 +Et3NH2=> Zn-HCOOH)
And I know than HI/RP will work.
But, Are any other routes to reduce the aminoalcohol to the aminoalkane?
Thanks for your time.
jep, i dont know but i have a feeling that a tosylester will work here to give good, maybe quantitative in a CTH?Sadly reactions are rarely driven to completion by feelings....

But, Are any other routes to reduce the aminoalcohol to the aminoalkane?

Until here all is fine, but now i need alternatives for the primary amine from the Aminoalcohol
2Electro S
Condensation could be done more OTC (cat. Al(OH)3, Al2O3)
 Or Zinc/ammonium formate???
Or Zinc/ammonium formate???thanks for the reply. Is my problem perhaps that I have been using water to dissolve my mercuric chloride?
A bucket seems so ghetto.
http://www.erowid.org/archive/rhodium/clandestine/bucket/index.html <-- like such?
is it simply for the extra head room to enable dumping the entire lot of al in at once? makes me nervous.